Inleiding
Het VEXAS-syndroom is een ontstekingsaandoening met manifestaties in het hele lichaam en wordt vaak in een laat stadium gediagnosticeerd. Het wordt gekenmerkt door hematologische afwijkingen en inflammatie. VEXAS is een acroniem voor vacuolen, E1 enzym, X-gebonden, auto-inflammatoir en somatisch. Er is sprake van een verworven somatische mutatie van het ubiquitine activerend enzym 1 (UBA1) gen, gelegen op het X-chromosoom (1). Mutaties in UBA1 belemmeren een goede intracellulaire afbraak van eiwitten, waardoor inflammatiepaden in gang worden gezet en een onbeheersbare ontstekingsreactie ontstaat (2). De overgrote meerderheid van de patiënten is man (omdat het om een X-gebonden ziekte gaat), er zijn enkele gevallen beschreven van het VEXAS-syndroom bij vrouwen, bijvoorbeeld bij monosomie van het X-chromosoom of het Turner syndroom. De huidige geschatte incidentie van het VEXAS-syndroom bedraagt 1 op de 4.269 witte mannen van 50 jaar en ouder in de Verenigde Staten (1).
Er is geen Tenth Revision (ICD-10) richtlijn voor het VEXAS-syndroom. Beck et al. hebben de volgende criteria gesteld waaraan moet worden voldaan om te spreken van het VEXAS-syndroom:
- UBA1-mutatie
- Klinische symptomen in ten minste twee orgaansystemen en ook afwijkende laboratoriumwaarden zoals anemie en/of trombocytopenie (3).
Patiënten met de diagnose VEXAS-syndroom lopen een verhoogd risico op morbiditeit en mortaliteit. De slechte uitkomst is het gevolg van een gebrek aan vroege herkenning en diagnose, progressie van de (hematologische) ziekte, een gebrek aan effectieve en efficiënte behandelingsmethoden en het oplopen van infecties (4).
Pathofysiologie
Bij het VEXAS-syndroom faalt het normale ubiquitine-proteasoom systeem. Het ubiquitine-proteasoom systeem bestaat uit twee stappen. De eerste stap is ubiquitinatie waarbij ubiquitine aan een af te breken eiwit wordt gekoppeld met behulp van drie enzymatische reacties. De tweede stap omvat de herkenning en afbraak van dit eiwit door het proteasoom (de-ubiquitinatie). Bij de mutatie in het UBA1 gen bij het VEXAS-syndroom wordt een pathologische isoform, UBA1c, gesynthetiseerd. Dit leidt tot stapeling van eiwitten, cel stress en ongecontroleerde inflammatie (5).
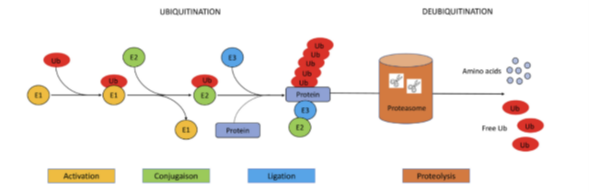
Ubiquitinatie en de-ubiquitinatie (5).
Symptomen
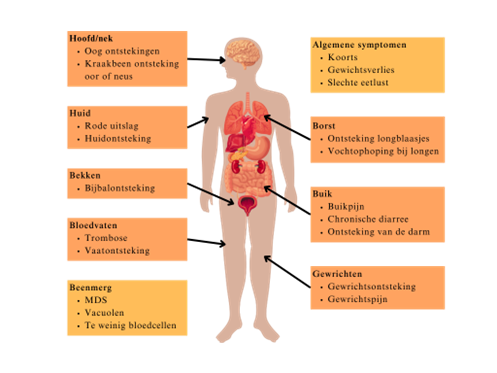
De eerste klinische uitingen van het VEXAS-syndroom manifesteren zich vaak tussen de 50 en 65 jaar. Deze uitingen zijn verspreid over het hele lichaam. Deze symptomen zijn vaak veelzijdig en kunnen verschillende orgaansystemen aantasten, waaronder de longen, de ogen, het kraakbeen, het gastro-intestinale systeem, het beenmerg, de bloedsomloop en de huid. De meest voorkomende symptomen zijn koorts, inflammatoire huiduitslag, respiratoire symptomen, veneuze trombose, artritis en vasculitis. Het myelodysplastisch syndroom komt in 30-63% van de gevallen voor (2).
Verschillende mutaties van het UBA1 gen zijn geassocieerd met verschillen in klinische presentatie van het VEXAS-syndroom. Deze variaties benadrukken het belang van genetische screening bij het diagnosticeren en behandelen van VEXAS-patiënten.
- p.Met41Val-mutatie:
- Lagere frequentie van chondritis
- Vaker een hoog CRP
- Hogere frequentie van MDS
- p.Met41Leu-mutatie:
- Een verhoogde vatbaarheid voor neutrofiele dermatose
- Vaker een mildere ziekte en een betere 5-jaarsoverleving in vergelijking met de andere 2 mutaties.
- p.Met41Thr-mutatie:
- Inflammatoire oogziekte (6)
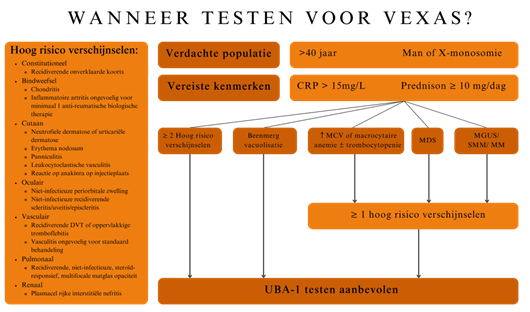
Uit onderzoek is onderstaand test algoritme ontwikkeld met een hoge positieve detectie ratio. Hoewel het VEXAS-syndroom veel minder vaak voorkomt bij vrouwen zijn ze wel meegenomen in het model. Met het huidige onderzoek is het niet bekend of de klinische presentaties bij mannen en vrouwen verschillen (2).
Differentiaaldiagnose
- Sweet syndroom en ander neutrofiele dermatosen
- Kan op basis van VEXAS-syndroom voorkomen, het sluit elkaar niet uit
- Polychondritis recidivans
- Poly Arteritis Nodosa (PAN)
- Giant Cell Arteritis (GCA)
- ANCA geassocieerde vasculitis
- MDS geassocieerde inflammatoire ziekten
- Kan op basis van VEXAS-syndroom aanwezig zijn, het sluit elkaar niet uit
- De ziekte van Behçet
- Inflammatoire darmziekten (IBD)
- Inflammatoire arteritis
- Macrofaag activatie syndroom (MAS)
Diagnostiek
Geadviseerd aanvullend onderzoek (met passende uitkomsten):
- Perifeer bloedonderzoek
- Verhoogd CRP
- Verhoogde pro inflammatoire cytokinen (5)
- Verhoogd MCV/ macrocytaire anemie (90%–100%) (2)
- Trombocytopenie (45%–69%)
- Monocytopenie (50%)
- Neutropenie (
- Next Generation Sequencing (NGS) voor het aantonen van een verworven UBA1 mutatie en additionele extra mutaties in andere genen (bloed of beenmerg)
- De UBA1-mutatie heeft vooral betrekking op substitutie van ATG-nucleotiden (Exon 3, codon 41). Er worden verschillende mutaties gezien, meest voorkomende varianten zijn (5i):
- 121A>G; p.Met41Val
- 121A>C; p.Met41Leu
- 122T>C; p.Met41Thr
- Beenmergonderzoek & chromosoomanalyse (2)
- UBA1-mutatie met mosaïcisme
- Hypercellulair beenmerg (90%)
- Granulocytaire hyperplasia met linksverschuiving (90%)
- Vergrote myeloïde/erytroïde ratio (M:E >4:1 bij 72%)
- Myelodysplastisch syndroom
- Vacuolen (niet altijd aanwezig)
- Chromosomen vaak niet afwijkend
- Moleculaire diagnostiek (verworven UBA1 mutatie)
- De UBA1-mutatie heeft vooral betrekking op substitutie van ATG-nucleotiden (Exon 3, codon 41). Er worden verschillende mutaties gezien, meest voorkomende varianten zijn (5i):
Behandeling
Er is nog geen richtlijn voor de behandeling van VEXAS-syndroom. Op dit moment is de behandeling gebaseerd op twee processen: het ingrijpen op de inflammatoire en immuun-paden of ingrijpen op de gemuteerde cellen. Vooral deze laatste strategie is veelbelovend, omdat hiermee de ziekte-load aantoonbaar afneemt en patiënten bij goede respons op den duur kunnen stoppen met de behandeling.
In de eerste groep heb je de volgende therapieën:
- Glucocorticosteroïden/prednisolon: veel patiënten gebruiken voor diagnose al prednisolon om de verschillende symptomen te bestrijden. Vaak wordt het nog langdurig gebruikt naast andere behandelingen. Over het algemeen zijn doseringen van 20-40 mg/dag nodig (7).
- Targeted synthetic DMARDs/JAK remmers (baricitinib, ruxolitinib): vooral ruxolitinib laat goede resultaten zien. Deze middelen hebben vaak een steroïde-sparende werking (8).
- Tocilizumab/anti-IL-6: werkt initieel goed om inflammatie te onderdrukken. Effect werkt na enige tijd uit. Het risico op infectieuze complicaties is vergroot. Vaak werkt tocilizumab voor ongeveer 8 maanden, totdat er een nieuwe behandeling moet worden gestart (7).
- Anakinra/anti-IL-1: voor patiënten met refractaire neutrofiele dermatose. Kan daarbij wel leiden tot ernstige cutane reacties die de behandeling lastig maken (7).
Onder behandelingen die aangrijpen op de UBAI gemuteerde cellen vallen:
- Hypomethyleerders (azacitidine): in eerste instantie werd dit alleen bij patiënten met MDS bij VEXAS-syndroom gebruikt, maar ondertussen ook steeds vaker bij patiënten zonder MDS. Het zorgt ervoor dat patiënten significant minder steroïden hoeven te gebruiken. Bij de overgrote meerderheid van de patiënten daalt de Variant allele frequency (VAF) van de UBA1 mutatie en soms zelfs tot zeer lage waarden zodat het middel gestopt kan worden en de ziekte rustig blijft (8, 9).
- Stamceltransplantatie: een allogene hematopoëtische stamceltransplantatie (HSCT) lijkt op dit moment de enige curatieve behandeling voor het VEXAS-syndroom. Niet elke patiënt is geschikt voor een allogene HSCT, vanwege de hoge risico’s op morbiditeit en mortaliteit. Het is belangrijk om de beslissing voor wel of geen allogene HSCT goed af te wegen en met de patiënt te bespreken (5). Bij progressieve cytopenieën of andere hoog-risico kenmerken, falen van andere behandelingen, sterke afhankelijkheid van prednison of aanwezigheid van andere klonale mutaties (e.g. DNMT3A, TET2), zou een stamceltransplantatie moeten worden overwogen (7).
Vooralsnog worden niet alle bovenstaande behandelingen vergoed. Voor het gebruik van azacitidine is een add-on aanvraag ingediend. Ruxolitinib wordt voor deze indicatie niet vergoed.
Sommige patiënten hebben ook andere ziekten die veroorzaakt worden door het VEXAS-syndroom. Op basis van deze andere ziekte is er soms wel een geregistreerde indicatie voor aanvullende behandelingen (zoals reumatoïde artritis).
Literatuurlijst
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease . New England Journal of Medicine. 2020 Dec 31;383(27):2628–38.
- Koster MJ, Lasho TL, Olteanu H, Reichard KK, Mangaonkar A, Warrington KJ, et al. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. Vol. 99, American Journal of Hematology. John Wiley and Sons Inc; 2024. p. 284–99.
- Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated with VEXAS Syndrome in a Clinical Population. JAMA. 2023 Jan 24;329(4):318–24.
- Zhang Y, Dong X, Wang H. VEXAS Syndrome—Review. Glob Med Genet. 2023 Sep;10(03):133–43.
- Khitri MY, Hadjadj J, Mekinian A, Jachiet V. VEXAS syndrome: An update. Vol. 91, Joint Bone Spine. Elsevier Masson s.r.l.; 2024.
- Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients*. British Journal of Dermatology. 2022 Mar 1;186(3):564–74.
- Koster MJ, Samec MJ, Warrington KJ. VEXAS Syndrome – A Review of Pathophysiology, Presentation, and Prognosis. Vol. 29, Journal of Clinical Rheumatology. Lippincott Williams and Wilkins; 2023. p. 298–306.
- Goring S, Horton J. Treatment Options for VEXAS Syndrome. 2022.
- Aalbers AM, van Daele PLA, Dalm VASH, Valk PJM, Raaijmakers MHGP. Long-term genetic and clinical remission after cessation of azacitidine treatment in patients with VEXAS syndrome. HemaSphere 2024. 8; e129