Introductie
Een normaal eosinofielengetal in het perifeer bloed is 0,1 tot 0,5 109/l. De WHO-classificatie onderscheidt de volgende graderingen van eosinofilie:
- Mild: bovengrens normaal tot 1,5 × 109/l
- Moderate: 1,5-5,0 × 109/l
- Severe: >5,0 × 109/l
Hypereosinofilie (HE) wordt gedefinieerd als een eosinofielengetal > 1,5 × 109/l in het perifere bloed. De volgende vormen worden onderscheiden:
- erfelijke vorm/familiare vorm
- primair klonaal/neoplastisch (HEN)
- vorm van onbekende betekenis/unknown significance (HEUS)
- secundair/reactief (HER)
De familiare vorm van HE is een autosomaal dominante afwijking (5q 31-33). Bij de geboorte is deze al aanwezig en is asymptomatisch. De vorm kan symptomatisch worden en/of overgaan naar een andere vorm. Bij de HEN is er onderliggend een klonale afwijking of maligniteit die de eosinofilie veroorzaakt. De HEUS is een asymptomatische vorm met onbekende origine en is een diagnose per exclusionem.
Secundaire vormen van eosinofilie zijn reactieve expansies met een grote diversiteit aan oorzaken. Er zijn diverse medicamenteuze en infectieuze oorzaken. Daarnaast kan atopie of een systeemziekte zoals SLE en eosinofiele granulomatose met polyangiitis (EGPA) de verklaring zijn. Daarnaast ligt de oorzaak soms in solide maligniteiten (voornamelijk hoofd-halstumoren) of hematologische maligniteiten (myeloïd [CML, AML, CMML, CEL, mastocytose] en lymfoid [voornamelijk T-cel en Hodgkin]). Er wordt dan een overvloed aan eosinofiele groeifactoren geproduceerd (IL-3, IL-5, GM-CF).
Eosinofilie kan lijden tot schade en/of disfunctie van eigenlijk alle orgaansystemen. De huid is het vaakst aangedaan bij ca. 70% van de patiënten, gevolgd door pulmonaal (44%), gastro-intestinaal (38%) en cardiaal (20%). De symptomen waarmee patiënten zich presenteren zijn hierdoor zeer verschillend. Vermoeidheid (26%), hoesten (24%), dyspnoe (16%), myalgie en angio-oedeem (beide 14%), huidrash en koorts (12%) en rhinitis (10%) zijn de meest voorkomende.
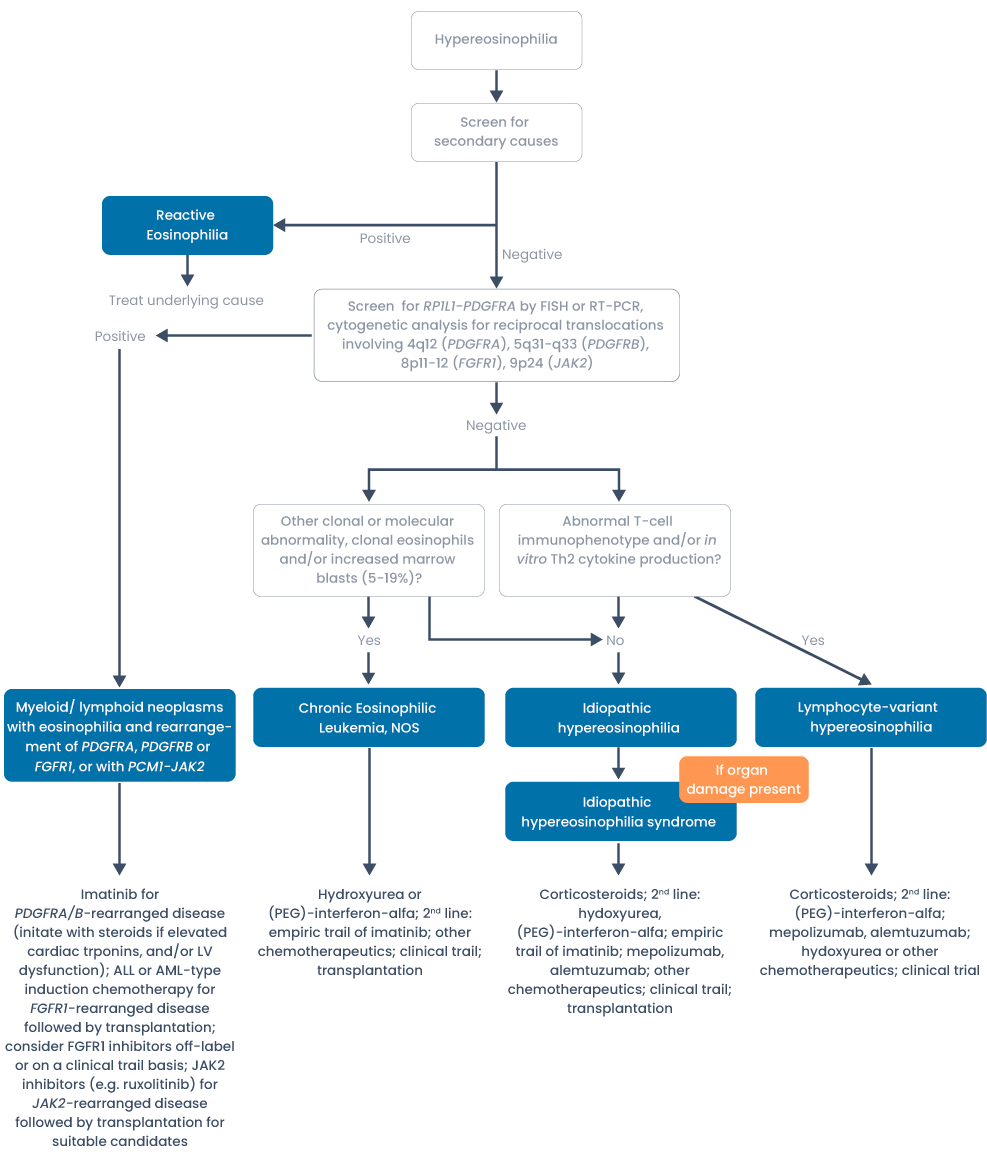
Het hypereosinofiel syndroom (HES) bestaat uit een heterogene groep aandoeningen met allen een verschillende etiologie, klinische manifestaties, behandelopties en prognose. De diagnose HES kan worden gesteld als er sprake is van hypereosinofilie (HE) én orgaanschade gerelateerd aan de HE, waarbij eosinofiele gastro-enteritis en eosinofiele pneumonie zijn uitgesloten. Er wordt onderscheid gemaakt in 3 subtypes HES.
Suptypes van hypereosinofiel syndroom
Myeloproliferatief Hypereosinofiel syndroom (M-HES)
Vaak betreft het jonge mannen met een agressief beloop van de ziekte. De meerderheid van deze patiënten heeft het fusiegen van Fip1-like 1 (FIP1L1) en platelet-derived growth factor receptor alpha (PDGFRA) op chromosoom 4q12; een gain of function-deletie waardoor een continu actief tyrosine kinase ontstaat, resulterend in expansie van eosinofiele populatie. Ook mutaties in de zeldzamere PDGFRB, FGFR1 en PCM1-JAK2 veroorzaken klonale eosinofilie die kan leiden tot M-HES.
Om de diagnose M-HES te stellen moet een van bovenstaande mutaties aantoonbaar zijn. Bij het ontbreken van een bepalende mutatie kan de diagnose gesteld worden als het klinisch beeld past bij een myeloproliferatieve aandoening (anemie, trombocytopenie, splenomegalie) én ≥ 4 van de volgende criteria aanwezig zijn:
| Criteria voor M-HES (indien geen fusiegen is gedetecteerd) |
| Dysplastische eosinofielen in handdif |
| Serum vitamine B12 > 1000 pg/ml |
| Anemie en/of trombocytopenie |
| Hepatosplenomegalie |
| Beenmerg cellulariteit > 80% |
| Myelofibrose |
| Spindle shaped mestcellen in het beenmerg |
Volgens de WHO-classificatie is er dan sprake van een myeloïde of lymfoide neoplasie met eosinofilie met rearrangement van PDGFRA, PDGFRB, FGFR1 of PCM1-JAK2.
Lymfatische variant HES (L-HES)
In L-HES wordt de eosinofilie veroorzaakt door een verhoogde productie van eosinofiele groeifactoren, voornamelijk interleukine-5 (IL-5), door geactiveerde T-lymfocyt populaties. Deze abnormale, klonale T-celpopulaties hebben een atypisch patroon van surface markers. Er zijn verschillende immunofenotyperingen bekend. De meest voorkomende is CD3-/CD4+. Daarnaast komt CD3+/CD4-/CD8- (immature T-cellen) met regelmaat voor.
De huid is het vaakst aangedaan bij L-HES patiënten, gevolgd door weke delen/lymfeklieren. Maar wederom kunnen eigenlijk alle orgaansystemen betrokken zijn. De meerderheid van de patiënten heeft een verhoogd serum IgE. Consensuscriteria voor de diagnose L-HES zijn er nog niet. De aanwezigheid van een geïsoleerde T-celkloon wordt nog niet voldoende geacht voor de diagnose. Indien er wel een aberrante T-celkloon is, maar géén weefselschade, dan valt dit onder de secundaire vormen van HE en is er geen sprake van HES.
Idiopatische HES (I-HES)
Dit is een diagnose per exclusionem. Er dient sprake te zijn van HES, en alle primaire en secundaire oorzaken van hypereosinofilie, M-HES en L-HES dienen uitgesloten te zijn. De diagnose is typisch in de 6e decade van het leven.
| Diagnostische criteria voor I-HES |
| 1. Eosinofilie > 1,5 × 109/l én geassocieerde weefselschade |
| 2. Niet voldaan aan de WHO-criteria voor: BCR-ABL1 CML, PV, ET, PMF, CNL, CMML en BCR-ABL1 negatieve CMML |
| 3. Geen rearrangement van: PDGFRA, PDGFRB of FGFR1, geen fusie van: PCM1-JAK2, ETV6 JAK2, of BCR-JAK2 fusion |
| 4. Blasten: perifeer < 2% en beenmerg < 5% |
| 5. Geen klonale cytogenetische of moleculaire afwijking (Cave CHIP kan uitzondering zijn, zeker bij ouderen) |
| 6. Sluit L-HES en M-HES uit |
Chronische Eosinofielen Leukemie not otherwise specificed (CEL-NOS)
Bij dit ziektebeeld wordt een hypereosinofilie gezien en voldaan aan onderstaande criteria. Typisch is presentatie in de 4e decade.
| Diagnostische criteria voor CEL-NOS |
| 1. Eosinofilie > 1,5 × 109/l én geassocieerde weefselschade |
| 2. Niet voldaan aan de WHO-criteria voor: BCR-ABL1 CML, PV, ET, PMF, CNL, CMML en BCR-ABL1 negatieve CMML |
| 3. Geen rearrangement van: PDGFRA, PDGFRB or FGFR1, geen fusie van: PCM1-JAK2, ETV6-JAK2, of BCR-JAK2 fusion |
| 4. Blasten: perifeer < 20% en beenmerg < 20%, geen AML-criteria |
| 5. Aanwezigheid van clonale cytogenetische en/of moleculaire afwijking (Cave CHIP kan uitzondering zijn, zeker bij ouderen) of blasten: perifeer > 2% en beenmerg > 5% |
| 6. Sluit L-HES en M-HES uit |
Orgaangebonden hypereosinofilie
Dit zijn aandoeningen waarbij een enkel orgaan is betrokken, gecombineerd met eosinofilie > 1,5 x 109 óf zeer duidelijke orgaaninfiltratie van eosinofielen. Deze beelden vallen niet onder HES. Voorbeelden zijn de gastro-intestinale HE en pulmonaire eosinofilie.
Overzicht van hypereosinofiele syndromen
| HES-variant | Subtype/type afwijking | Klinische en labkenmerken |
| Myeloïd
(M-HES en CEL-NOS) |
PDGFRB en FGFR1 rearrangement
JAK-2 puntmutaties en translocaties CEL NOS |
Serum vit B12 verhoogd
Anemie en trombopenie Hepato- en/of splenomegalie Circulerende myeloide voorlopers |
| Del 4q12 -> FIP1L1-PDGFRA fusie | Evt. verhoogde serum tryptase en mestcelafwijkingen | |
| PDGFRA of PDGFRB rearrangement-geassocieerde ziekte vrijwel uitsluitend bij mannen | ||
| L-HES | Aberrante IL-5 producerende T-cellen
Bijv. CD3-CD4+ T-cel-geassocieerde ziekte |
Duidelijke huidafwijkingen (plaques, erytroderma, urticaria)
Polyclonale hypergammaglobulinemie Meestal benige lymfoproliferatieve aandoening, maar kan progressie naar T-cel NHL vertonen |
| Fam. HES | Gelokaliseerd op 5q 31-33 | Asymptomatische eosinofilie vanaf de geboorte (autosomaal dominant)
Progressie naar andere vormen is mogelijk |
| Orgaangebonden HE | Bijv. eosinofiele gastro-intestinale ziekte, chronische eosinofiele pneumonie, e.d. | Perifere eosinofilie met eosinofiele infiltraten en geassocieerde klachten/ symptomen in een orgaan |
| Specifieke syndromen met HE | Bijv. episodisch angio-oedeem met eosinofilie, eosinofiele granulomatose met polyangiitis (EGPA), e.a. | Eosinofilie bij een andere onderliggende oorzaak die geassocieerd is met eosinofilie |
Diagnostiek
Anamnese
Gezien de zeer diverse betrokkenheid van orgaansystemen is een uitgebreide tractus anamnese van uiterst belang om mogelijke pathologie te kunnen detecteren. Daarnaast is een reisanamnese van belang om gerichte diagnostiek naar (parasitaire) infecties in te kunnen zetten gedurende de work-up. Tevens dient het medicatie-, kruiden- en supplementgebruik van de afgelopen tijd gedetailleerd uitgevraagd te worden.
Lichamelijk onderzoek
Een volledig internistisch lichamelijk onderzoek dient uitgevoerd te worden. Specifieke aandachtspunten hierbij zijn eventuele hepatosplenomegalie, lymfadenopathie en mogelijke orgaanbetrokkenheid. Tevens is een (oriënterend) neurologisch onderzoek geïndiceerd. Een WHO-performance is nuttig ter beoordeling van functioneren en snelheid die nodig is in de diagnostiek (klinisch/poliklinisch).
Laboratoriumonderzoek
- Bloedbeeld met handdif (afwijkende morfologie van eo’s is niet richtinggevend)
- Chemie met nierfunctie, leverproeven, cardiale enzymen (TropT, NTproBNP), CK, CRP
- Serum tryptase (mastocytose, ook verhoogd bij PDGFRA/PDGFRB fusies)
- Vitamine B12 (vaak verhoogd bij M-HES)
- Serum immunoglobulines (specifiek IgE, vaak verhoogd bij L-HES)
- Auto-immuundiagnostiek: ANA en bij hoge verdenking op EGPA ook ANCA
- TARC (of IL-5 spiegel) (bij lymfadenopathie onder de verdenking van L-HES)
- Cortisol (bij verdenking op eventuele onderliggende bijnierschorsinsufficiëntie)
Aanvullend onderzoek
- Medische microbiologie: Onderzoek naar parasitaire infectie (serologie naar zeker Toxocariasis, Strongyloides stercoralis en verse feces indien klachten of positieve serologie). Op basis van anamnese eventueel uit te breiden.
Tevens voor pulmonaire aspergillose. Daarnaast diagnostiek naar EBV, soms wordt L-HES hierdoor gedreven. - Longfunctie (op indicatie).
- ECG en echocardiografie of MUGA-scan (zeker voor start imatinib!).
- CT-thorax/abdomen (bij verdenking op lymfadenopathie en of hepato-/splenomegalie).
- Weefselbiopsie, gericht op afwijkingen gevonden bij anamnese/lichamelijk onderzoek/diagnostiek.
- Beenmergonderzoek (ip bij persisterende eosinofilie > 1,5 × 109/l):
- cytomorfologie
- flowcytometrie
- cytogenetica (FISH gericht op onderstaande mutaties)
- moleculaire diagnostiek (PCR of NGS) gericht op:
-
-
- FIP1L1-PDGFRA fusiegen (chr.4; alleen aantoonbaar met (RT-)PCR of FISH);
- PDGFRB–fusies, met name met ETV6, zoals bij de t(5;12)(q33;p13), en andere fusiecombinaties vanuit PDGFRB op 5q33 (aCML, CMML-beelden);
- 8p11-breuken (FGFR1, fibroblast growth factor receptor 1 betrokkenheid) (MDS/Mypro-beelden);
- JAK2-mutatie;
- BCR-ABL;
- KITD816V-mutatie;
- Klonaliteitsanalyse T-celreceptor, m.n. CD2+CD3–CD4+CD25+ TCRαβ–HLA-DR+CD45RO+, m.a.w. klonale, geactiveerde, premaligne memory-T-cellen.
-
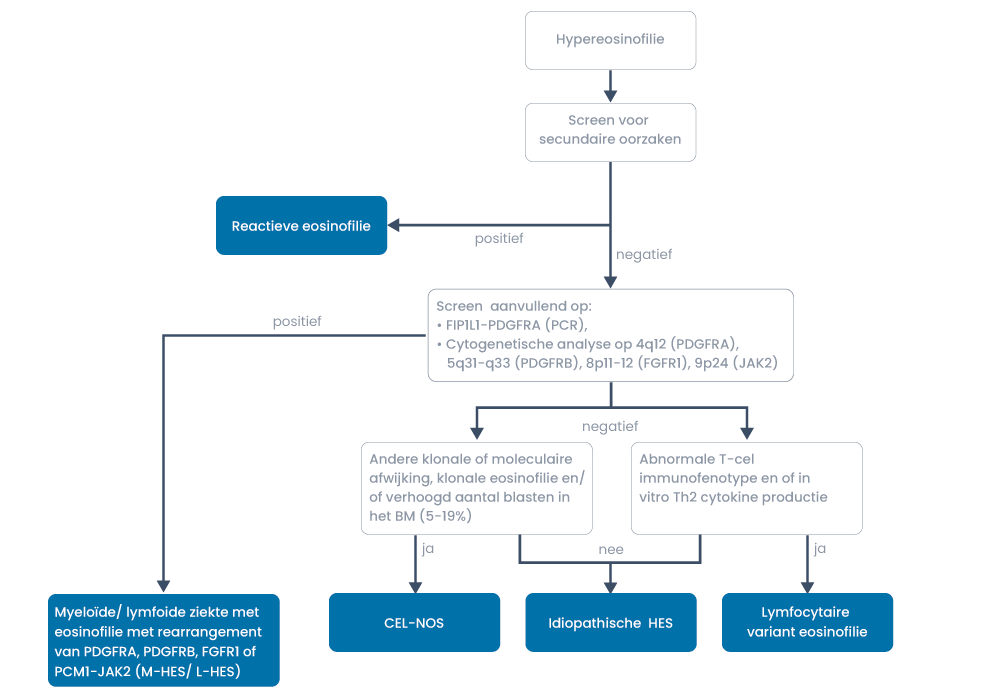
Flowdiagram diagnostiek
Prognose
M-HES: van oudsher was het risico op (cardiaal) overlijden zeer groot. De mediane overleving was 9 maanden met een 3-jaar-overleving van slechts 12%. Een retrospectieve studie van Legrand (2013) liet zien dat patiënten die behandeld worden met imatinib wegens een FIP1L1-PDGFRA-positieve ziekte een goede prognose hebben. Slechts 1 van de 44 patiënten was overleden bij een mediane follow-up van 52 maanden.
L-HES: dit is een meer indolent verlopende ziekte. Echter hebben patiënten een verhoogd risico op het ontwikkelen van een T-cellymfoom of Sezary-syndroom. Accumulatie van cytogenetische veranderingen (vooral partieel 6q, 10p-deleties en trisomy 7) zijn een verhoogd risico voor progressie naar een lymfoom.
I-HES: De overall 5-jaarsoverleving ligt rond de 80%. Echter is er een hoge variabiliteit bij de onbekende en vermoedelijke heterogene origine. Patiënten overlijden aan hartfalen, CVA’s en complicaties van langdurig steroïdengebruik.
CEL-NOS: De mediane overleving is 15-22 maanden. De meeste patiënten overlijden aan progressie naar AML of beenmergfalen.
Behandeling
Filter trials en protocollen voor dit ziektebeeld
Bij de verdenking op een parasitaire oorzaak van de eosinofilie, behandel dit laagdrempelig (i.o.m. een infectioloog) en beoordeel het effect op de eosinofilie.
Acute noodzaak tot behandeling
Hematologische emergencies die acute behandeling behoeven zijn:
– Extreme eosinofielenlevels (> 100 × 109/l);
– Tekenen van leukostase (bij leukocyten getal 50 × 109/l);
– Potentieel levensbedreigende complicaties van HE (ernstig hartfalen, trombo-embolieën [vaak cardiaal/arterieel], pulmonaal falen).
Start met hoge dosis steroïden IV (1 mg/kg prednisolon of 1 gram methylprednisolon), eventueel met ivermeticine indien risico op Strongyloides. Je verwacht > 50% daling van het eosinofielengetal binnen 24 uur. Indien deze therapie onvoldoende aanslaat kan overwogen worden om daarnaast te starten met:
– Imatinib (400 mg), zeker als het een M-HES beeld is
– Vincristine (1 à 2 mg/m2)
– Hydroxyurea (500-1000 mg/dag, snel op te hogen naar 2000 mg/dag)
– Cyclofosfamide, vooral bij EGPA-beeld
Eerste lijn (niet acuut)
M-HES met PDGFRA of PDGFRB rearrangements: imatinib. Start dosering 400 mg/dag. Typisch verbetering in 1 à 2 weken. Eventueel dosis reduceren. Start steroïden (prednison 1-2 mg/kg/dag) bij verminderde linkerventrikelfunctie op echo cor en/of verhoogd TropT en/of NT-proBNP.
M-HES met JAK2-mutatie/translocatie: JAK-2 inhibitors (ruxolitinib) of hydroxyureum.
M-HES met FGFR1 rearrangements: inductie-chemotherapie (AML-based) met als doel allogene stamceltranplantatie.
M-HES beeld zonder mutatie: corticosteroïden (prednison 20-60 mg 1dd).
L-HES: corticosteroïden (prednison: 20-60 mg 1dd).
I-HES: corticosteroïden (prednison: 20-60 mg 1dd).
CEL-NOS: hydroxyureum of peginterferon-alfa.
Tweede lijn en/of steroïdesparend
M-HES met PDGFRA of PDGFRB rearrangements: andere TKI (dasatinib, sorafenib, nilotinib) of allogene stamceltransplantie.
M-HES met JAK2-mutatie/translocatie: geen advies; erbij of switch naar peginterferon-alfa.
M-HES met FGFR1 rearrangements: allogene stamceltranplantatie.
M-HES beeld zonder mutatie: imatinib op proef (eventueel op te hogen naar 800 mg 1dd), hydroxyureum of peginterferon-alfa.
L-HES: voorkeur is behandeling met mepolizumab (monoclonaal antilichaam tegen IL-5). Een alternatief is interferon. Tevens kan chemotherapie gebruikt worden (chloorambucil, etoposide, vincristine, cladribine in combinatie met cytarabine). Verder worden in de literatuur alemtuzumab (monoclonaal antilichaam tegen CD-52) en ciclosporine als mogelijkheden beschreven (echter wordt alemtuzumab hiervoor niet vergoed).
I-HES: Hydroxyureum (start 500-1000 mg/dag, op te hogen naar 2000 mg/dag bij tolerantie). Een alternatief is peginterferon-alfa. Bij onvoldoende effect zijn mepolizumab, alemtuzumab en chemotherapie zoals bij L-HES te proberen.
CEL-NOS: Eventueel op proef imatinib. Overweeg allogene stamceltransplantatie.
Studies lopen voor diverse subtypes met anti-IL-5-therapie (reslizumab en benralizumab) en met dexpramixpexole (onbekend werkingsmechanisme).
Bijzonderheden/specifieke aandachtspunten, ziekte-/behandelinggerelateerd
Het doel van de verschillende behandelingen is een reductie van het absolute eosinofielengetal en hierdoor symptomen te behandelen en verdere ziekteprogressie (orgaanschade) te voorkomen. Gedurende de behandeling dient naast het bloedbeeld ook orgaanfunctioneren vervolgd te worden. Cardiale fibrose is meestal niet reversibel, maar progressie kan met adequate therapie wel geremd of voorkomen worden.
Wees alert op het verhoogd risico op trombotische complicaties. Profylactische behandeling met antistolling wordt niet geadviseerd.
Follow-up
Bij intensieve behandeling wekelijks met bloedbeeldcontroles en vervolg van aangedane orgaansystemen. Indien patiënt stabiel is ingesteld op therapie en de eosinofilie onder controle is, kan dat worden uitgebreid naar uiteindelijk 1 à 2 keer per jaar.
Literatuurlijst
- Gotlib, J. World Health Organization-defined eosinophilic disorders: 2017 update on diagnosis, risk stratification, and management. Am J Hematol. 2017;92:1243–1259.
- Valent, P., Klion, A.D., Horny, H-P., et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130: 607-12.
- Klion, A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology 2018; 326-331
- Klion, A. How I treat hypereosinophilic syndromes. Blood. 2015;126(9):1069-1077
- Wang, S.A. The Diagnostic Work-Up of Hypereosinophilia. Pathobiology 2019;86:39–52
- Legrand F, Renneville A, Macintyre E, et al. The Spectrum of FIP1L1-PDGFRA-Associated Chronic Eosinophilic Leukemia: New Insights Based on a Survey of 44 Cases. Medicine (Baltimore) 2013.
- Roufosse F., Klion, A.D. Weller, P.F. (2022, 6 april), Hypereosinophilic syndromes: Clinical manifestations, pathophysiology, and diagnosis. UptoDate.com. geraadpleegd op 21-6-2022. =
- Roufosse F., Klion, A.D. Weller, P.F. Hypereosinophilic syndromes: Clinical manifestations, pathophysiology, and diagnosis. UptoDate.com. (2020, 24 november), geraadpleegd op 25-6-2022
- Up-to date