Windows en Linux
Houdt de toets ctrl ingedrukt en draai het muiswiel omhoog of omlaag
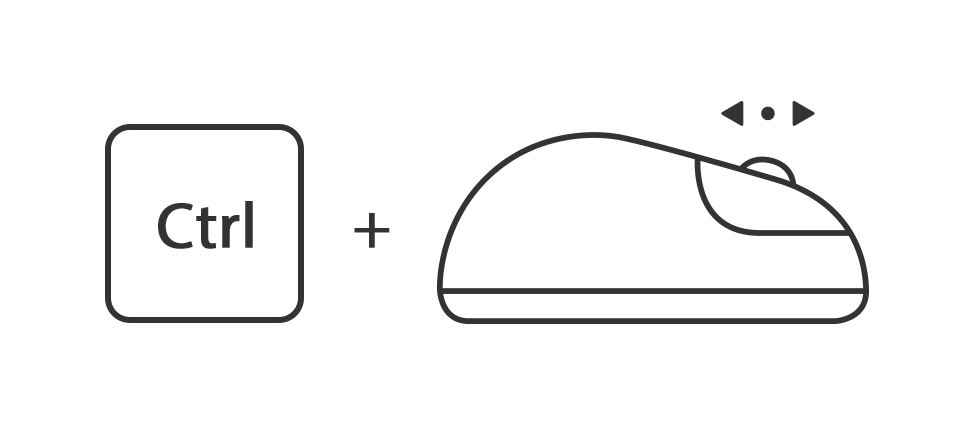
Of houdt de toets ctrl ingedrukt en druk tegelijk op de toets + of -
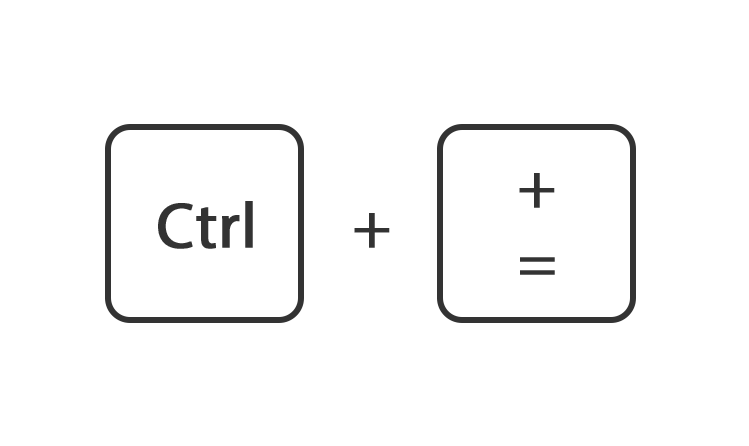
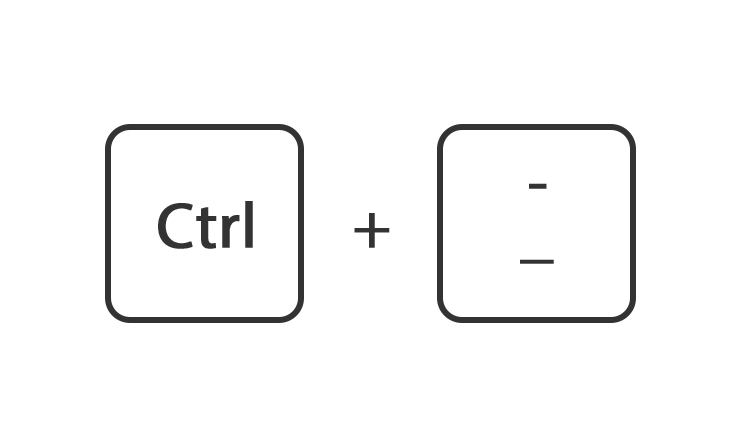
Of ga rechtsboven naar instellingen en gebruik de instelling zoom
Mac
Houdt de toets command ingedrukt en druk op de toets + of -
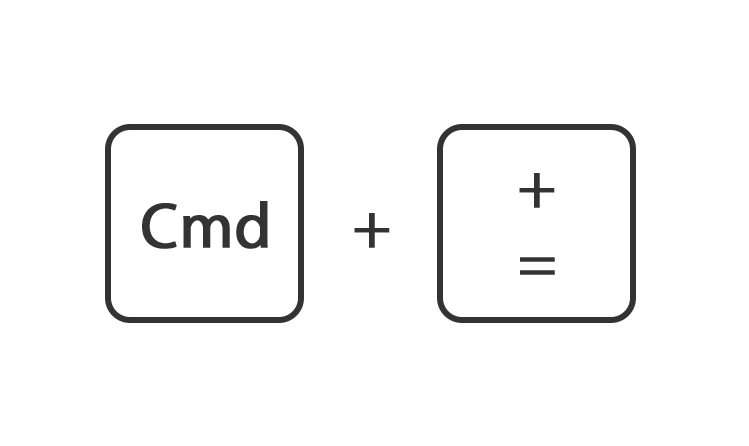
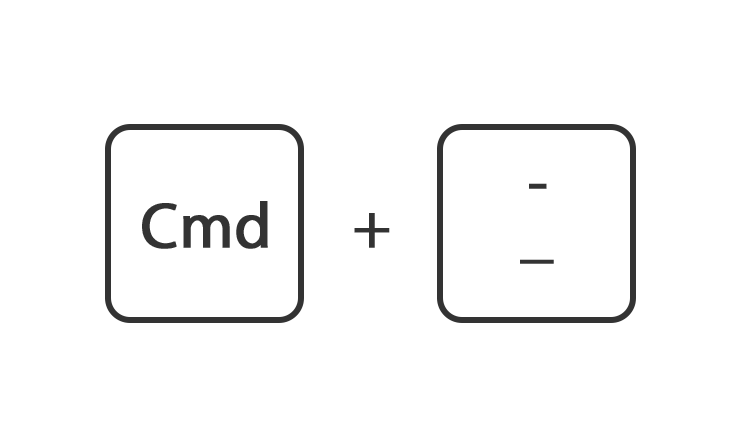
Of ga rechtsboven naar instellingen en gebruik de instelling zoom.