Wat is het VEXAS-syndroom?
Het VEXAS-syndroom is een zeldzame ontstekingsziekte die overal in het lichaam tot ziekteverschijnselen kan leiden. VEXAS is een afkorting dat de typische kenmerken van de ziekte beschrijft (vacuolen, E1-enzym, X-linked, auto-inflammatoir en somatisch). De oorzaak van het VEXAS-syndroom is een verandering in het erfelijk materiaal (DNA), een dergelijke verandering heet mutatie. De mutatie bij het VEXAS-syndroom heet de UBA1-mutatie. Deze afwijking ligt op het X-chromosoom. Dit is één van de twee geslachtschromosomen die elk persoon heeft. Een man heeft één X- en één Y-chromosoom, een vrouw heeft twee X-chromosomen (zie afbeelding).
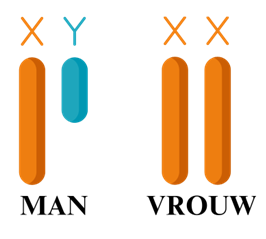
De ziekte ontstaat tijdens het leven (en is dus niet erfelijk) en komt vooral voor bij (oudere) mannen, doordat mannen maar één X-chromosoom hebben. Als het enige X-chromosoom de afwijking heeft, ontstaat de ziekte. Bij vrouwen is er dan nog een ander gezond X-chromosoom dat de functie kan overnemen. Door de mutatie werken bepaalde stoffen in het lichaam niet meer goed (1).
Hoe wordt het VEXAS-syndroom herkend, wat zijn de klachten (symptomen)
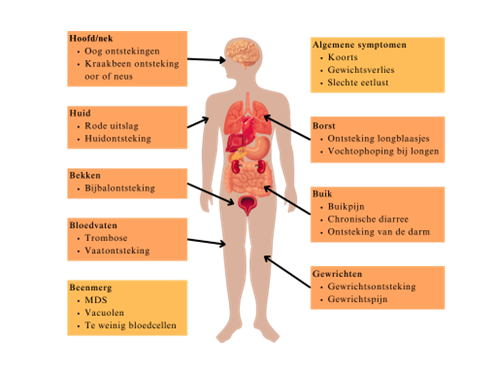
Mensen met het VEXAS-syndroom krijgen meestal tussen hun 50e en 65e last van de eerste klachten van deze ziekte. De klachten kunnen overal in het lichaam voorkomen en verschillen vaak per persoon. Zoals in de longen, darmen, nieren, ogen, gewrichten, huid en het bloed. De meest voorkomende klachten zijn: koorts, een rode huiduitslag, ademhalingsproblemen, stolsels in de bloedvaten, ook wel bekend als trombose, en pijn in de gewrichten. Een ernstige uiting van de ziekte is het myelodysplastisch syndroom (MDS). Dit is een stoornis in het beenmerg waardoor de productie van bloedcellen ernstig verstoord is. Dit komt bij ongeveer 30-63% van de patiënten voor (zie ook hoofdstuk MDS).
Op het plaatje hieronder zijn alle klachten (symptomen) per orgaansysteem weergegeven.
Bloed en beenmergonderzoek
Om de diagnose VEXAS-syndroom te kunnen stellen worden een aantal onderzoeken gedaan: bloedonderzoek en beenmergonderzoek. Artsen kunnen het VEXAS-syndroom bewijzen door het aantonen van de mutatie van het UBA1-gen (in bloed of beenmerg).
Bloedonderzoek: In het bloed kunnen verschillende afwijkingen gevonden worden die duiden op het VEXAS-syndroom. Bijvoorbeeld:
- Verhoogde ontstekingswaarden (CRP)
- Vergrote rode bloedcellen, wat macrocytose wordt genoemd
- Bloedarmoede (te weinig rode bloedcellen), dit wordt anemie genoemd
- Verlaagd aantal bloedplaatjes, dit wordt trombocytopenie genoemd
- Verlaagd aantal witte bloedcellen, dit wordt leukopenie genoemd
- De specifieke mutatie, zogenaamd UBA1-mutatie (5)
Beenmergonderzoek: Het beenmerg is het zachte weefsel binnen in je botten waar bloedcellen worden gemaakt. Met dit onderzoek wordt gekeken of er specifieke aanwijzingen zijn die duiden op het VEXAS-syndroom. Meer informatie over hoe dit onderzoek gaat is te vinden op:
https://hematologiegroningen.nl/patienteninformatie/verrichtingen/beenmergonderzoek/.
Afwijkingen die in het beenmerg gevonden kunnen worden zijn:
- Een te grote hoeveelheid cellen, wat hypercellulair beenmerg wordt genoemd
- Te veel afweercellen (granulocyten)
- Een verkeerde verhouding tussen de verschillende soorten beenmergcellen
- Myelodysplastisch syndroom (MDS), zie eerder of in het desbetreffende hoofdstuk
- Typerende kenmerken van het VEXAS-syndroom zoals vacuolen (blaasjes gevuld met vocht) in de cellen in het beenmerg
- Specifieke mutaties in het UBA1-gen, aangetoond door middel van moleculaire diagnostiek van bloed of beenmerg (7)
Behandeling van VEXAS
Op dit moment is er nog geen vastgestelde richtlijn voor de behandeling van het VEXAS-syndroom. Er zijn (nog) geen medicijnen specifiek gericht tegen dit ziektebeeld. De behandeling richt zich momenteel op twee processen: het aanpakken van ontstekings- en immuunprocessen in het lichaam, of het uitschakelen van de cellen die afwijkend (gemuteerd) zijn. We verdelen de medicijnen dus in 2 groepen. Soms kunt u een combinatie van deze groepen krijgen voor de behandeling van uw specifieke situatie.
Groep 1:
- Prednisolon
- Baracitinib en ruxolitinib: beide JAK remmers
- Anakinra: een anti-IL-1
- Tocilizumab: een anti-IL-6
Deze medicijnen zorgen ervoor dat de ontsteking die door de ziekte ontstaat minder actief is en u minder klachten heeft, maar kunnen VEXAS-syndroom niet genezen. Prednisolon werkt vaak goed, maar heeft bijwerkingen als botontkalking, spierklachten, gewichtstoename en infecties. Om die reden worden er vaak andere medicijnen naast prednisolon gebruikt, om de benodigde hoeveelheid prednisolon zo laag mogelijk te houden.
Groep 2:
- Azacitidine een hypomethyleerders
- Stamceltransplantatie
Een stamceltransplantatie is een genezende behandeling, maar niet iedereen kan een stamceltransplantatie krijgen omdat dit een ingrijpende, zware behandeling is met potentieel veel en ernstige bijwerkingen. Meer informatie over een stamceltransplantatie is te vinden op:
https://hematologiegroningen.nl/patienteninformatie/verrichtingen/stamceltransplantatie-introductie/.
Azacitidine is een nieuwere behandeling die erg goede resultaten laat zien en misschien zelfs genezend zou kunnen zijn. Hiernaar is meer onderzoek nodig.
Belangrijk te weten is, doordat de ziekte heel recent (sinds 2020) is ontdekt, dat er nog maar weinig onderzoeken over de behandeling zijn en dat daarom ook niet alle behandelingen door de verzekering worden betaald.
Prognose
Omdat het VEXAS-syndroom een recent ontdekte ziekte is, is er nog veel onbekend. Ook over de prognose. De ernst van de ziekte hangt af van de ernst van de symptomen en hoe lang die er al zijn. Als de ziekte slecht reageert op verschillende behandelingen, is de prognose slechter. De ziekte is progressief en kan leiden tot overlijden, maar de huidige behandelingen slaan in de meeste gevallen (tijdelijk) aan en leiden tot minder klachten of soms zelfs een langdurige fase zonder ziekteverschijnselen.
Conclusie/ samenvatting
Het VEXAS-syndroom is een zeldzame ontstekingsziekte veroorzaakt door de UBA1-mutatie op het X-chromosoom. Het treft vooral mannen (tussen de 50-65 jaar) en kan verschillende symptomen veroorzaken, zoals koorts, huiduitslag, ademhalingsproblemen, klontjes in het bloed (trombose) en gewrichtspijn.
Om het VEXAS-syndroom te diagnosticeren, worden bloed- en beenmergonderzoeken uitgevoerd. Bloedonderzoek kan afwijkingen zoals verhoogde ontstekingswaarden en de aanwezigheid van de UBA1-mutatie aan het licht brengen. Beenmergonderzoek kan aanvullende kenmerken van het syndroom en de UBA1-mutatie bevestigen. Deze diagnostische tests helpen artsen bij het herkennen van het VEXAS-syndroom en het plannen van passende behandelingen voor patiënten.
Op dit moment is er nog geen specifieke behandeling vastgesteld voor het VEXAS-syndroom. Medicijnen voor de behandeling worden verdeeld in twee groepen, waarbij soms een combinatie wordt gebruikt. Over de prognose is nog veel onbekend. Behandelingen slaan in de meeste gevallen (tijdelijk) aan, maar het blijft een ernstige en progressieve ziekte die kan leiden tot overlijden.
Literatuurlijst
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease . New England Journal of Medicine. 2020 Dec 31;383(27):2628–38.
- Koster MJ, Lasho TL, Olteanu H, Reichard KK, Mangaonkar A, Warrington KJ, et al. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. Vol. 99, American Journal of Hematology. John Wiley and Sons Inc; 2024. p. 284–99.
- Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated with VEXAS Syndrome in a Clinical Population. JAMA. 2023 Jan 24;329(4):318–24.
- Zhang Y, Dong X, Wang H. VEXAS Syndrome—Review. Glob Med Genet. 2023 Sep;10(03):133–43.
- Khitri MY, Hadjadj J, Mekinian A, Jachiet V. VEXAS syndrome: An update. Vol. 91, Joint Bone Spine. Elsevier Masson s.r.l.; 2024.
- Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients*. British Journal of Dermatology. 2022 Mar 1;186(3):564–74.
- Koster MJ, Samec MJ, Warrington KJ. VEXAS Syndrome – A Review of Pathophysiology, Presentation, and Prognosis. Vol. 29, Journal of Clinical Rheumatology. Lippincott Williams and Wilkins; 2023. p. 298–306.
- Goring S, Horton J. Treatment Options for VEXAS Syndrome. 2022.