Inleiding
De porfyrieën zijn zeldzame, hoofdzakelijk erfelijke, maar soms verworven aandoeningen waarbij stoornissen in de biosynthese van het haem bestaan ten gevolge van (partiële) deficiënties van enzymen die bij de verschillende stappen van de haemsynthese betrokken zijn. Door deze enzymdeficiënties, vaak in combinatie met externe uitlokkende factoren, treedt toxische accumulatie van tussenmetabolieten (porfyrines) op wat leidt tot neurotoxiciteit en/of huidafwijkingen door fotosensibiliteit.
De porfyrieën kunnen op verschillende manieren worden ingedeeld. Zo kan er onderscheid worden gemaakt tussen primaire en secundaire porfyrieën. De primaire porfyrieën zijn de erfelijke porfyrieën, waarvan porphyria cutanea tarda (PCT) overigens meestal verworven is. Secundaire porfyrieën zijn andere aandoeningen, zoals loodintoxicatie, alcoholische hepatitis, en sideroblastische anemie, die de haemsynthese beïnvloeden en daardoor verhoogde concentraties van de tussenproducten van de haemsynthese kunnen veroorzaken. De term porfyrie wordt in de praktijk voornamelijk gereserveerd voor de primaire porfyrieën. De indeling in primair en secundair is vooral nuttig voor het inzicht dat veel andere aandoeningen het porfyrinemetabolisme kunnen beïnvloeden en dus ook afwijkende uitslagen van porfyrinetesten kunnen geven.
Een andere classificatie is gebaseerd op de plaats waar het grootste deel van de overmaat aan porfyrines bij een specifieke enzymdeficiëntie wordt geproduceerd, namelijk de lever of het beenmerg. Haemsynthese vindt namelijk hoofdzakelijk in het beenmerg en de lever plaats (zie onder). Op die manier onderscheidt men hepatische en erytropoïetische porfyrieën.
In de klinische praktijk wordt meestal de indeling acuut versus cutaan gehanteerd. Deze classificatie is niet optimaal omdat twee vormen van porfyrie (HCP en PV) zowel acute als cutane symptomen kunnen geven. Daarnaast zijn er cutane porfyrieën die acute huidsymptomen veroorzaken (XLP en EPP). De indeling is echter relevant aangezien met name het herkennen van een acute porfyrie-aanval belangrijk is. Zo’n aanval kan namelijk levensbedreigend zijn indien deze niet adequaat wordt behandeld.
Haemsynthese
Haem is het eindproduct van acht opeenvolgende enzymatische stappen en bestaat uit een complex van protoporfyrine-IX en ijzer (zie figuur 1). Haem wordt gesynthetiseerd in alle metabool-actieve cellen in het lichaam die mitochondriën bevatten, maar voornamelijk in het beenmerg (85% van het totaal), als bouwsteen voor hemoglobine, en in de lever (ca. 15%), als onderdeel voor bepaalde enzymen die betrokken zijn bij oxydatie, celdifferentiatie, eiwitsynthese, hydroxylering en de detoxificatie van exo- en endogene stoffen in de lever. Dit zijn voornamelijk de cytochroom-P450-enzymen.
Haem wordt in acht reactiestappen uit glycine en succinyl-coenzym-A (succinyl-CoA) gesynthetiseerd, waarbij voor elke reactie een specifiek enzym nodig is (figuur 1). Het snelheidsbepalende enzym in de haemsynthese is het eerste enzym: delta-aminolevulinezuur-synthase (ALA-synthase of ALAS). Van ALAS bestaan twee iso-enzymen: ALAS1 en ALAS2 waarvan de genen op twee verschillende chromosomen liggen (resp. chromosoom 3 en het X-chromosoom). ALAS1 heeft een sleutelrol bij de regulatie van de haemsynthese in de lever. Haem reguleert ALAS1 via negatieve feedback: een hoge haemconcentratie in de lever leidt tot afname van de synthese van ALAS1, wat weer resulteert in afname van de haemsynthese. Omgekeerd leidt een toegenomen vraag naar haem in de lever tot upregulatie van ALAS1. Dit laatste gebeurt bijvoorbeeld wanneer in de lever meer cytochroom-P450(CYP)-enzymen geproduceerd worden door gebruik van CYP-inducerende middelen zoals bepaalde medicatie of alcohol.
ALAS2 wordt alleen geproduceerd in erytroblasten en speelt samen met ijzer een rol bij de regulatie van de haemsynthese in het beenmerg.
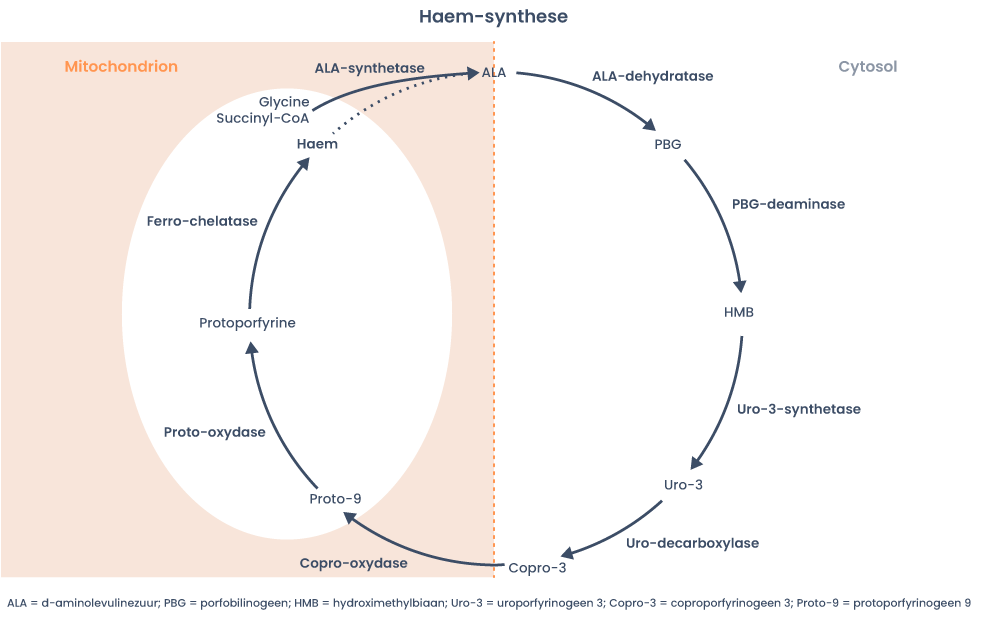
Figuur 1: De haemsynthese (naar Andersson, 1997)
Primaire porfyrie
Er zijn acht verschillende porfyrieën bekend. Van de enzymdefecten verantwoordelijk voor de porfyrieën zijn de genlocaties bekend. Van alle enzymen die betrokken zijn bij de haemsynthese zijn ziekte-veroorzakende genmutaties beschreven, behalve van ALAS1. Mutaties in het ALAS2-gen veroorzaken X-chromosoom gebonden sideroblastische anemie (XLSA) (‘loss-of-function’-mutaties) of X-chromosoom gebonden protoporfyrie (‘gain-of-function’-mutaties). XLSA wordt vaak ingedeeld bij de porfyrieën, maar in tegenstelling tot de andere porfyrieën gaat XLSA niet gepaard met cutane of (acute) neuroviscerale symptomen. XLSA wordt gekenmerkt door lichte hypochrome microcytaire anemie en ernstige ijzerstapeling. Daarom is XLSA eigenlijk geen porfyrie.
De meest voorkomende porfyrieën zijn acute intermitterende porfyrie (AIP), porfyria cutanea tarda (PCT) en erytropoietische porfyrie (EPP).
Vier porfyrieën erven autosomaal dominant over met een geringe penetrantie (zie tabel 1). Bijna alle aangedane individuen hebben een heterozygote mutatie. De enzymactiviteit is hierdoor ongeveer 50% van normaal, wat meestal voldoende is voor een normale haemsynthese. Het grootste deel (80-90%) van de getroffen personen zal dan ook geen klachten of problemen met de haemproductie ondervinden. Verreweg de meeste patiënten met erfelijke aanleg voor een van deze vormen van porfyrie zullen nooit klachten krijgen (± 90%). Klachten kunnen echter wel optreden bij blootstelling aan uitlokkende factoren, zoals bepaalde medicatie, alcoholgebruik, laag-calorisch dieet of infecties. Activatie van de haemsynthese via extra activatie van ALAS1 resulteert op dat moment in toxische accumulatie van porfyrines. Homozygote vormen van de autosomaal dominante vormen zijn extreem zeldzaam en manifesteren zich in een veel ernstiger, eerder beginnend en chronischer ziektebeeld. Bij autosomaal recessieve vormen van porfyrie is er chronische accumulatie van porfyrines doordat de resterende enzymactiviteit veel lager is. Zoals boven vermeld worden de porfyrieën in de praktijk meestal ingedeeld op basis van klinische verschijnselen (acute of cutane) en de plaats van de haem- en porfyrineproductie in het lichaam (beenmerg of lever). Deze indeling in acute en cutane porfyrieën is niet geheel sluitend, aangezien porfyrieën zowel acute als cutane verschijnselen kunnen vertonen (zie tabel 1 en figuur 2).
| Porfyrie | Deficiënt enzym | Klinische verschijnselen | Overerveringspatroon |
| X-gebonden protoporfyrie | ALAS2 | Cutaan | X-chromosoom gebonden |
| ADP = ALAD-deficiëntie-porfyrie | ALA-dehydratase (ALAD) | Acuut | Autosomaal recessief |
| AIP = Acute intermitterende porfyrie | PBG-deaminase (PBGD) | Acuut | Autosomaal dominant |
| CEP = Congenitale erytropoëtische porfyrie | Uro-3-synthetase (UROS) | Cutaan | Autosomaal recessief |
| PCT = Porphyria cutanea tarda | Uro-decarboxylase (UROD) | Cutaan | Autosomaal dominant* |
| HCP = Hereditaire coproporfyrie | Copro-oxidase (CPOX) | Acuut + cutaan | Autosomaal dominant |
| PV = Porphyria variegata | Protoporfyrinogeen-oxidase (PPOX) | Acuut + cutaan | Autosomaal dominant |
| EPP = Erytropoëtische protoporfyrie | Ferrochelatase (FECH) | Cutaan | Autosomaal dominant |
* Verminderde UROD-activiteit bij PCT is meestal verworven. Een homozygote of compound heterozygote vorm van UROD-deficiëntie is zeer zeldzaam. Deze vorm wordt hepato-erytropoietische porfyrie (HEP) genoemd.
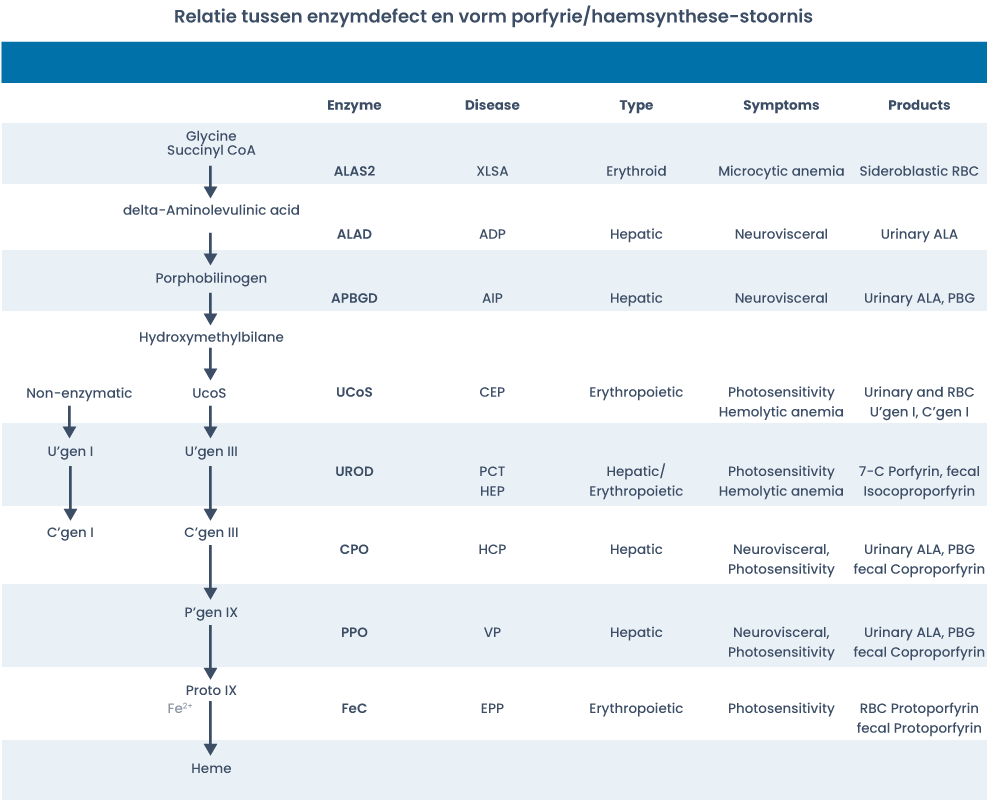
Figuur 2: De relatie tussen enzymdefecten en vormen van porfyrie/haemsynthese-stoornisen
Acute porfyrie
De acute porfyrieën zijn hepatische porfyrieën. De meest voorkomende vorm van acute porfyrie is de acute intermitterende porfyrie (1:20.000).
Bij een aanval van acute porfyrie is er een overproductie van δ-ALA en PBG, porfyrine-voorstadia die neurotoxisch zijn. Hierdoor treden er neuroviscerale symptomen op (zie onder bij kliniek). Tijdens deze aanvallen wordt een verhoogde uitscheiding van δ-ALA en PBG in de urine gevonden, waardoor de urine bij hoge concentraties van PBG en bij langdurige UV-expositie rood/bruin kleurt (het Griekse ‘πορΦνρα’ betekent ‘purper’). De pathogenese van de neurologische dysfunctie bij de acute-porfyrie-aanval is nog niet opgehelderd. Er zijn aanwijzingen dat δ-ALA neurotoxisch is, maar ook dat het de receptor-activiteit van γ-aminoboterzuurreceptor (GABA), een belangrijke neurotransmitter, kan beïnvloeden. Ander onderzoek suggereert dat een tekort aan haem in neurale cellen een rol speelt in de pathogenese.
Kliniek
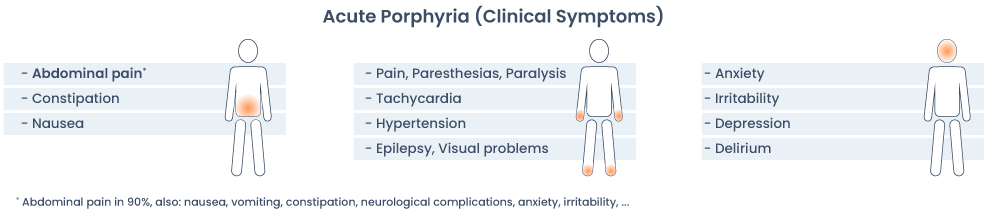
De klinische manifestaties van acute porfyrie bestaan uit acute aanvallen en soms chronische verschijnselen als de acute aanval niet op tijd wordt behandeld of als de aanvallen gepaard gaan met ernstige neurologische uitval. Het klassieke beeld bestaat uit recidiverende aanvallen van hevige koliekachtige buikpijn, met daarnaast een scala van neurologische klachten. Tussen de aanvallen door kunnen lange perioden van remissies voorkomen.
Intermitterende neurologische symptomen (zie ook figuur 3):
- Autonome neuropathie
- Koliekpijn, obstipatie, braken, paralytische ileus
- Tachycardie, hypo- of hypertensie, urineretentie, koorts
- Perifere neuropathie (vooral motorisch)
- proximale spierzwakte van de extremiteiten, quadriplegie, heesheid, respiratoire insufficiëntie door paralyse van de ademhalingsspieren
- CZS-veranderingen
- blindheid, emotionele labiliteit, psychosen, tonisch-clonische insulten, coma, visusstoornissen
- hyponatriemie (t.g.v. SIADH door hypothalamus-aantasting of door ondervulling t.g.v. braken)
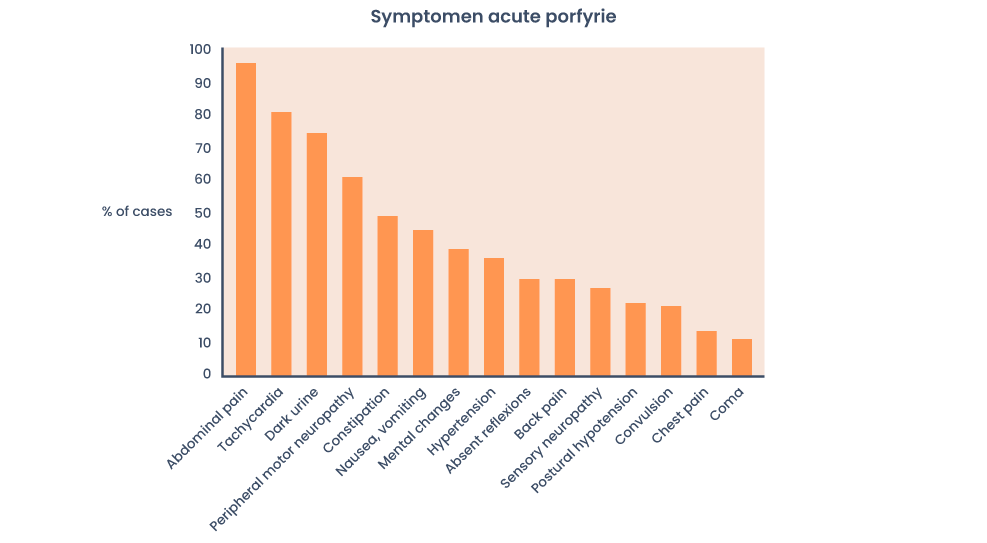
Figuur 3: Frequentie van symptomen tijdens een acute aanval van porfyrie
Predisponerende factoren
Het enzym ALA-synthase (ALAS) bepaalt de snelheid van de haemsynthese. Dit enzym kan worden geactiveerd door exogene factoren. Hierdoor ontstaat er een toename van toxische metabolieten en treedt exacerbatie van het ziektebeeld op.
Belangrijke exogene uitlokkende factoren zijn:
- hormonen
- alcohol
- vasten/afvallen/katabole toestand
- infecties
- roken
- medicatie; zie www.porphyria-europe.com voor een lijst van medicatie
Diagnostiek
Door oxidatie van tussenproducten van de haemsynthese die in overmaat in de urine worden uitgescheiden, ontstaat een typische donkerbruine tot purperachtige kleur. Centraal staat de bepaling van het δ-ALA- en het PBG-gehalte in de urine, waarden zijn 3-10x verhoogd tijdens een aanval.
Bij verdenking van een porfyrie-aanval: bepaal altijd cito δ-ALA- en het PBG-gehalte in een portie urine. Elk laboratorium in Nederland kan deze bepaling cito uitvoeren.
In de remissiefase kunnen de toxische metabolieten nauwelijks verhoogd zijn (zie ook figuur 4).
Therapie
Algemeen ondersteunende therapie
Doel is uitlokkende exogene factoren uit te schakelen, en katabolie en stress te voorkomen. Hierdoor treedt afname van de δ-ALA-synthase-activiteit en van de accumulatie van toxische metabolieten op.
- Start direct met glucose 10% infuus 2 l/24 uur. Glucose remt de δ-ALA-synthaseactiviteit.
- Bij ontstaan van hyperglykemie behandel met insuline, ga niet over op NaCl-infuus.
- Infecties dienen prompt behandeld te worden.
- Zorg voor adequate calorische intake, evt. sondevoeding (koolhydraatrijk).
- Check medicatie die mogelijk aanval kan hebben uitgelokt en staak die (bijv. hormonen, diclofenac, metoclopramide, sulfonamide-antibiotica, zie voor lijst www.porphyria-europe.com).
- Bij koorts: start paracetamol.
- Bij pijn: paracetamol of morfine.
- Bij braken : chloorpromazine.
- Hypertensie: propanolol, atenolol.
- Convulsies: diazepam i.v., clonazepam (corrigeer evt. hyponatriemie).
- Bij acute adrenerge crisis, ernstige hypertensie, encefalopathie, insulten: Magnesiumsulfaat.
- Wees alert op het ontstaan van hyponatriemie in kader SIADH, dit kan neurologische complicaties verergeren. Bij SIADH: vochtbeperking. Overweeg bij neurologische symptomen snellere correctie met hypertoon zout.
Specifieke therapie
Bij ernstige aanval (ernstige maagdarmklachten, cardiovasculaire klachten en alle neurologische uitingsvormen)
- Humaan hemine (Normosang, Recordati Rare Diseases) zo snel als mogelijk. Hematine veroorzaakt in de lever een repressie van het snelheidsbepalende enzym δ-ALA-synthetase (ALAS).
- Starten bij aanval, dan meest effectief, duur afhankelijk van symptomen; ten minste 4 dagen, bij langduriger beloop aanval continueren.
- Dosering: bij lichaamsgewicht > 65 kg 1 ampul 250 mg normosang 1dd1 (anders ~3 mg/kg/dag).
- Toediening: normosang in 100 ml NaCl 0,9% in 30 min of in 100 ml albumine 20% (geeft minder flebitis).
Preventie en vervolg
Bij de behandeling van patiënten met acute porfyrie is het van belang dat patiënten, ter voorkoming van een aanval, zelf een lijst van niet-toegestane geneesmiddelen bezitten. Sites met dergelijke lijsten zijn:
Indien een aanval van acute porfyrie is gediagnosticeerd is verwijzing naar regionaal of landelijk expertise centrum te adviseren (landelijk: Erasmus MC Porfyrie Centrum Rotterdam). Hier wordt verdere typering verricht en familiescreening besproken.
Cutane porfyrie
De cutane porfyrie zijn meestal erytropoitische porfyrieën. Door aanwezigheid van porfyrinen in de huid ontstaat bij blootstelling aan zonlicht (en soms kunstlicht) een huiddermatose. Doordat accumulatie van porfyrinen ook in de erytrocyten optreedt, kan soms hemolyse optreden in de huidcapillairen.
Huidafwijkingen die optreden op de zon-geëxposeerde delen zijn: bullae, oedeem, jeuk, lichenificatie, hypertrichose, hyperpigmentatie en chronische ulcera.
Porfyria cutanea tarda (PCT) is de meest voorkomende vorm van cutane porfyrie (prevalentie ca. 1 op 10.000). PCT ontstaat door verminderde UROD-activiteit. Dit is meestal verworven. Circa 20% van de patiënten met PCT heeft een heterozygote UROD-genmutatie die autosomaal dominant overerft. De meeste mensen met een heterozygote UROD-genmutatie ontwikkelen echter geen PCT, doordat een dergelijke mutatie de UROD-activiteit met circa 50% verlaagt, terwijl een UROD-activiteit van < 20% pas klinische verschijnselen van PCT veroorzaakt. Voor het bereiken van zo’n lage activiteit is de aanwezigheid van externe factoren/andere aandoeningen, zoals alcoholgebruik, virale hepatitis, roken, oestrogenengebruik of de aanwezigheid van HFE-mutaties, noodzakelijk. Een homozygote of compound heterozygote UROD-genmutatie is zeer zeldzaam (slechts ongeveer 40 beschreven gevallen wereldwijd). Dit veroorzaakt hepato-erytropoietische porfyrie (HEP). De kliniek van PCT en HEP bestaat hoofdzakelijk uit chronische fotosensibiliserende bulleuze huidafwijkingen en leverenzymstoornissen (verhoogde transaminasen) zonder acute aanvallen.
Predisponerende factoren
- Zonlicht;
- Alcohol;
- Hormonen (oestrogenen);
- Medicatie; zie www.uq.edu.au/porphyria voor lijst van medicatie.
Diagnostiek
Met behulp analyse van urine, soms feces en bloed, kan de diagnose gesteld worden (zie figuur 4).
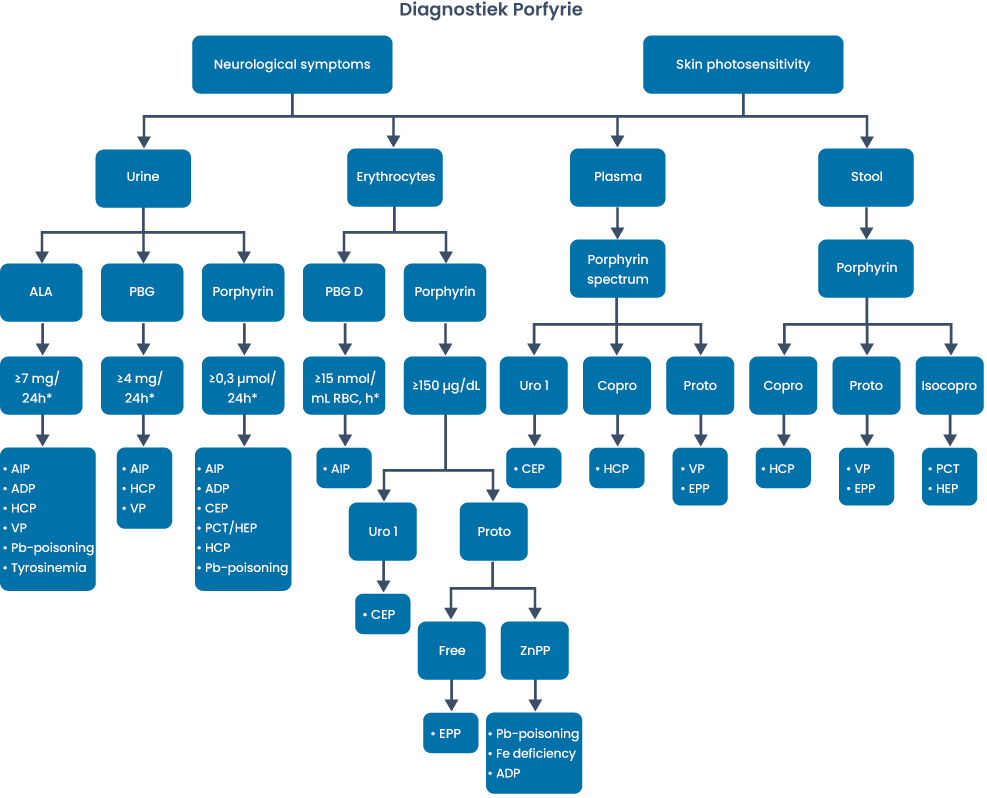
Figuur 4: De diagnostiek van porfyrie
Therapie/beleid
- Voorkomen expositie UV-straling, sunblock factor > 20
- Staken alcoholgebruik
- Evt. chloroquine 2 × 125 mg
- Evt. colestyramine (Questran) (onderbreking enterohepatische circulatie van protoporfyrine)
- Beta-caroteen 1 × 150 mg (bij EPP)
- Bij Porfyria cutanea tarda screenen op mutaties in HFE-gen, HIV, hepatitis C
- Flebotomie tot ferritine < 20
Secundaire porfyrie
Verhoogde concentraties van tussenproducten in de haemsynthese komen voor bij meerdere ziektebeelden. Dit wordt veroorzaakt door een verminderde uitscheidingscapaciteit van lever of nier en door directe remming van bepaalde enzymen. Zo wordt de werking van ALAD geremd bij loodintoxicatie en hereditaire tyrosinemie type 1. Het excretiepatroon wijkt af van dat van de primaire porfyrieën doordat veelal maar 1 soort porfyrine is verhoogd. Bij de volgende ziektebeelden wordt verhoogde pofyrinen-excretie gevonden:
- Loodintoxicatie – delta-aminolevulinezuur
- Hereditaire tyrosinemie type 1 – delta-aminolevulinezuur
- Chronische nierziekten – uroporfyrine I
- Bloedziekten: anemie, leukemie, polycytemia vera – uroporfyrine I, coporfyrinen I
- Leverziekten: Gilbert, Dubin-Johnson en Rotor; levercirrose, alcoholintoxicatie – coporfyrinen I
Literatuurlijst
- Beukeveld, G.J.J., Wolthers, B.G. Laboratoriumdiagnostiek bij porfyrieen. Ned Tijdschr Klin Chem 1997; 22: 3-14.
- Jansen, J.J.W.M. en Huijgens, P.C. Porfyrie. In: Löwenberg, B., Ossenkoppele. G.J., Witte de, T., Boogaerts, M.A. (red.): Handboek Hematologie. Utrecht, De Tijdstroom, 2008.
- Kauppinnen, R. Seminar Porphyrias. Lancet 1997; 349: 1613-1617.
- Mccoll, K.E., Moore, M.R., Thompson, G.G., Goldberg, A. Treatment with hematin in acute hepatic porphyria. Q J Med 1981; 198: 161-74.
- Pischik E, Kaakov V, Kauppinen R. Is screening for urninary porphobilinogen useful; among patients with acute polyneuropathy or encephalopathy? J Neurol 2008;255:974-979
- Siersema, P.D., Wilson, J.H.P. De Porfyrieen. Ned Tijdschr Geneeskd 1989; 133: 2542-2547.
- Bottomley, S.S. Porphyria. In: Greer J.P. et al (Ed.): Wintrobe’s Clinical Hematology. Philadelphia, Lippincott Williams & Wilkins, 2009
- Harrison’s Principles of Internal Medicine, Twentieth Edition, McGraw-Hill Education, 2018
- Wiley J.S. & Moore M.R. Heme biosynthesis and its disorders: porphyrias and sideroblastic anemias. In: Hoffman, R. et al (Ed.): Hematology: Basic principles and practice. Philadelphia, Elsevier Inc., 2005