Wat is myelodysplastisch syndroom?
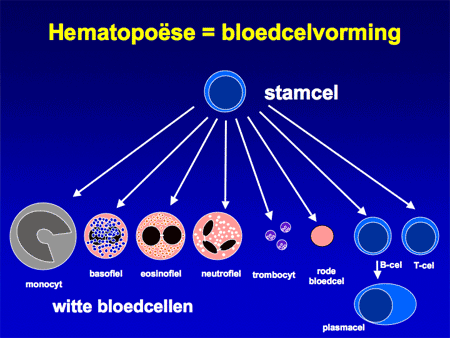
Myelodysplastisch syndroom (MDS) wordt ook wel myelodysplasie genoemd en staat voor een groep van beenmergstoornissen waarbij de productie van bloedcellen ernstig is verstoord. De normale bloedaanmaak speelt zich af in het beenmerg (zie ook Beenmergonderzoek) (dia 2 en 3). Daarbij worden de verschillende celsoorten aangemaakt, zoals de rode cellen (nodig voor zuurstoftransport; een tekort geeft bloedarmoede), witte cellen (leukocyten genaamd; er zijn veel verschillende soorten en ze zijn nodig voor de afweer tegen bacteriën, virussen en andere infectiebronnen) en bloedplaatjes (trombocyten genaamd, nodig voor de bloedstolling).

Dia 1
Dia 2
Dia 3
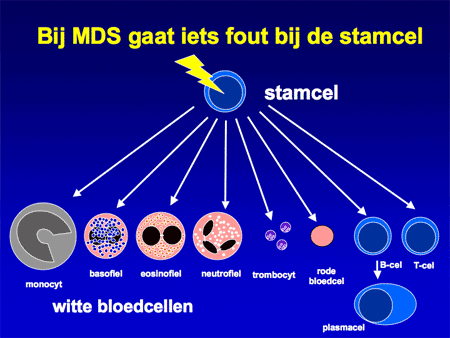
Bij MDS worden de bloedcellen niet goed aangemaakt. Het resultaat van deze gestoorde aanmaak zijn misvormde en niet goed uitgegroeide bloedcellen. Deze misvormingen worden dysplasie genoemd. Vandaar ook de naam: myelodysplasie (mergdysplasie). Door de slechte kwaliteit van de bij MDS geproduceerde bloedcellen wordt een belangrijk deel van deze bloedcellen al vernietigd voordat zij het beenmerg verlaten. Daardoor kan een tekort ontstaan aan deze bloedcellen. Een tekort aan rode cellen heet anemie (bloedarmoede), een tekort aan witte cellen heet leukocytopenie en een tekort aan trombocyten/bloedplaatjes heet trombocytopenie.
Oorzaken
In de meeste gevallen van MDS kan geen oorzaak voor de gestoorde aanmaak worden aangewezen. Aannemelijk is dat de aanmaakfout ligt in de bloed- en beenmergstamcel. Aangenomen wordt dat tijdens de vele delingen van beenmergcellen fouten zijn opgetreden, die uiteindelijk tot de ziekte MDS geleid hebben. Daarom is MDS ook typisch een ziekte van ouderen (ouder dan 60 jaar), hoewel de ziekte zeker ook kan voorkomen bij jongeren. Eén belangrijke oorzaak moet genoemd worden, namelijk de MDS die kan ontstaan na chemotherapie (de groep van celdodende medicijnen die bij de behandeling van kanker wordt gebruikt). Hoe tegenstrijdig dit ook lijkt: sommige celdodende middelen kunnen op hun beurt ook weer een kwaadaardige beenmergziekte, namelijk MDS of acute myeloïde leukemie (AML) veroorzaken.

Dia 4
MDS en acute leukemie
Bij MDS is niet alleen sprake van productie van gestoorde, niet goed functionerende bloedcellen, maar de voorlopercellen kunnen soms (bij ongeveer een derde van de patiënten) ook ontsporen en kwaadaardig worden. In het ergste geval kan zich dit ontwikkelen richting acute myeloïde leukemie (AML).
De kans of MDS acute leukemie wordt hangt sterk af van het type MDS (zie Indeling van MDS en de varianten). Sommige vormen van MDS ontsporen vrijwel nooit, andere veel vaker.
Hoe wordt MDS herkend?
Patiënten met MDS hoeven nauwelijks klachten te hebben. De afwijking wordt nogal eens bij toeval ontdekt bij een bloedonderzoek dat om een andere reden wordt verricht. Het ontbreken van klachten wordt verklaard door het feit dat de ziekte zeer langzaam kan ontstaan, waardoor het lichaam gaat wennen aan het tekort aan bloedcellen. Sommige patiënten echter hebben wel klachten en symptomen die veroorzaakt zijn door het tekort aan normale cellen. Door de bloedarmoede kan algemene zwakte en bleekheid met onverklaarbare, toenemende vermoeidheid gezien worden. Door het tekort aan witte cellen kunnen veelvuldig terugkerende kleine infecties en koorts ontstaan. Door het tekort aan bloedplaatjes ontstaan gemakkelijk blauwe plekken of bloedingen, bijvoorbeeld neusbloedingen, kleine huidbloedingen (kleine rode stipjes op het lichaam) of tandvleesbloedingen (bijvoorbeeld bij het tandenpoetsen). Gelukkig treden bloedingen niet vaak op, omdat de bloedplaatjes bij MDS meestal niet gevaarlijk laag zijn.

Dia 5
Beenmergonderzoek en chromosoomanalyse
Om de diagnose MDS te stellen zijn bloed- en beenmergonderzoek nodig. Het bloed en het beenmerg worden onder de microscoop beoordeeld op de aanwezigheid van – voor MDS typische – dysplastische afwijkingen (dia 7, 8 en 9). Daarnaast wordt het aantal blasten geteld. Blasten zijn normale gezonde voorlopercellen, maar kunnen ook een uiting van leukemie zijn: een teveel aan blasten wijst op een ontwikkeling richting acute myeloïde leukemie.
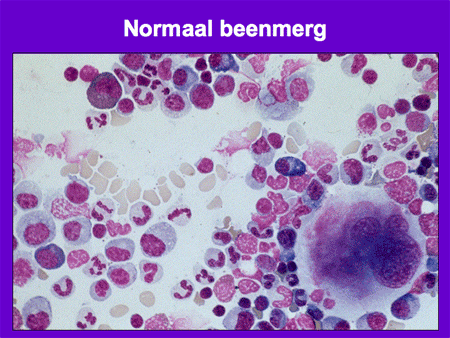
Dia 6
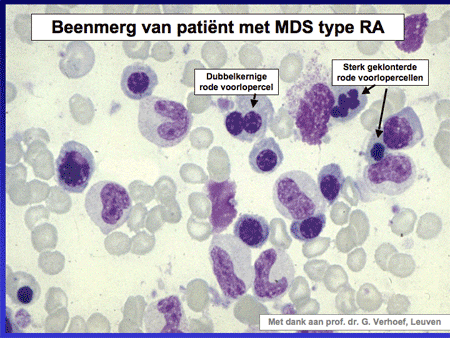
Dia 7
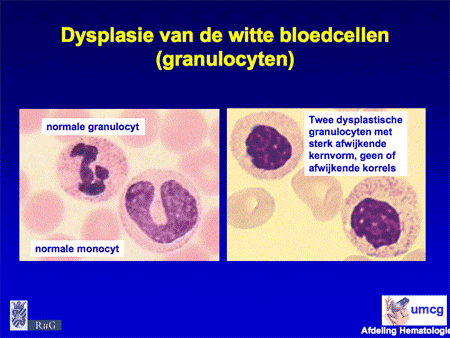
Dia 8
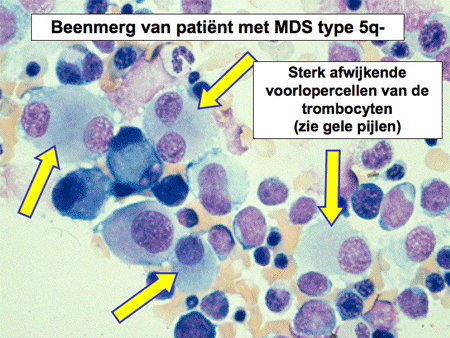
Dia 9. Beenmerg van patiënt met MDS type 5q-
Tot slot wordt het opgezogen beenmerg gekweekt om de chromosomen te analyseren (cytogenetisch onderzoek). Bij dit onderzoek wordt gekeken naar chromosoomafwijkingen in de afwijkende cellen, niet in de cellen van de rest van het lichaam. De aanname is immers dat alleen in de beenmergstamcellen afwijkingen zijn opgetreden, die mogelijk met chromosomenonderzoek aangetoond kunnen worden. De rest van de lichaamscellen is volledig normaal. Er is ook geen sprake van een erfelijke aandoening of iets dergelijks. Een chromosoomanalyse is van grote steun, omdat sommige afwijkingen heel karakteristiek bij MDS voorkomen en dan kunnen helpen de diagnose met meer zekerheid te kunnen stellen. Bovendien is er een subgroep MDS met een typische chromosoomafwijking (het 5q- syndroom; spreek uit: vijf-kuu-min), dat met een nieuw medicament (lenalidomide) vaak uitstekend te behandelen is.
MDS is niet altijd makkelijk te herkennen, omdat andere aandoeningen, zoals infecties, of gebruik van bepaalde medicamenten ook afwijkingen aan het beenmerg kunnen veroorzaken die erg op MDS kunnen lijken. In dat geval is het de moeite waard een paar maanden te wachten en het beenmergonderzoek te herhalen.
Indeling van MDS en de varianten
Het is gebruikelijk dit ziektebeeld in een aantal groepen in te delen die stuk voor stuk voorspellend zijn voor de kans op verergering richting acute leukemie. Deze indeling wordt overal in de wereld gebruikt. Naast de hoofdgroep MDS wordt een tweede groep onderscheiden die veel meer op een chronische, langzaam groeiende leukemie lijkt. De indeling en naamgeving is helaas erg complex met veel afkortingen. Ze wordt bepaald door de aan- of afwezigheid van anemie, leukopenie of trombopenie, het percentage blasten in het beenmerg, en de aanwezigheid van specifieke chromosoomafwijkingen.

Dia 11
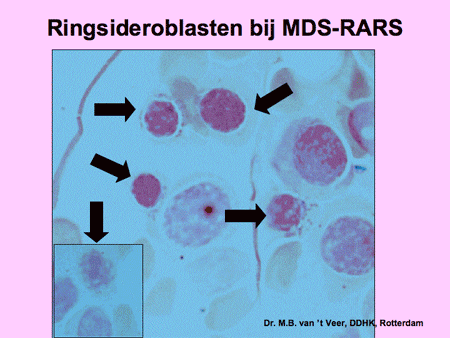
Dia 12
De indeling ziet er als volgt uit:
- Hoofdgroep van MDS:
-
Refractaire anemie (RA)
Dit type komt in ongeveer 20 tot 30 procent van de gevallen van MDS voor. De afwijkingen concentreren zich vooral in de aanmaak van de rode cellen. De meeste patiënten hebben bloedarmoede (anemie). Deze anemie reageert niet op bijvoorbeeld het toedienen van ijzertabletten, vandaar de naam ‘refractair’. Het aantal blasten in het beenmerg is laag. Zelden is er een ontwikkeling richting acute leukemie. De gemiddelde overlevingsduur bedraagt vele jaren.
-
Refractaire anemie met ringsideroblasten (RARS)
Het beeld is identiek aan MDS type RA, maar er worden extra afwijkingen waargenomen bij de rode bloedcellen. Het ijzer, dat normaal als een enkel korreltje opgeslagen wordt in de zich ontwikkelende rode bloedcel, is hier sterk toegenomen, waarbij een karakteristieke ring wordt gevormd. Deze cellen worden ‘ringsideroblasten’ genoemd. Ongeveer 2 tot 5 procent van de MDS-patiënten heeft deze vorm van MDS. De overlevingskansen zijn gelijk aan die van MDS (RA). Een enkele keer reageren patiënten goed op zeer hoge doses vitamine B6 (pyridoxine).
-
Refractaire cytopenie met multilineage dysplasie (RCMD)
Dit beeld lijkt erg op de refractaire anemie. Ditmaal zijn niet alleen de rode cellen, maar ook de witte cellen en/of bloedplaatjes bij het beeld betrokken. Het woord ‘cytopenie’ (een tekort aan cellen) geeft aan dat bij deze patiënten sprake kan zijn van een anemie en/of leukocytopenie en/of trombocytopenie.
-
Refractaire anemie met excess aan blasten (RAEB)
Bij deze vorm (die ongeveer 30 tot 35 procent van de MDS-patiënten betreft) komen meer abnormale primitieve bloedcellen voor. Het aantal blasten in het beenmerg, en soms ook in het bloed, is toegenomen (‘excess’). Ongeveer 40 procent van de patiënten met dit type MDS krijgt uiteindelijk een vorm van acute leukemie, meestal AML. Daarom zijn de overlevingskansen zonder therapie slechter.
-
MDS met 5q- afwijking
Deze zeldzame vorm van MDS wordt apart beschreven. Het betreft nogal eens oudere vrouwelijke patiënten met een bloedarmoede (anemie) en toename van trombocyten. In het beenmerg worden karakteristieke afwijkingen aan de megakaryocyten gezien, de voorlopercellen van de bloedplaatjes. De prognose wat betreft overleving is over het algemeen beter dan van de andere vormen van MDS. Ook is de kans op ontwikkeling richting acute myeloïde leukemie erg klein. Voor deze vorm van MDS is het middel lenalidomide beschikbaar.
-
- MDS lijkend op myeloproliferatie:
-
Chronische myelomonocytenleukemie (CMML c.q CMMoL)
Dit ziektebeeld wordt gerangschikt onder de subgroep MDS richting myeloproliferatie. Myeloproliferatie wil zeggen: toegenomen groei van beenmergcellen. Deze vorm werd vroeger tot de MDS gerekend, maar gedraagt zich meer als een chronische leukemie. Karakteristiek is er een toename van één bepaald type witte bloedcellen, namelijk de monocyten. Deze toename wordt in het bloed gezien (monocytosis) en in het beenmerg, met wederom soms een toename van blasten. Er is meestal een geringe verlaging van het aantal bloedplaatjes en er kan sprake zijn van anemie. Ook deze vorm kan uiteindelijk bij ongeveer 30 procent van de patiënten overgaan in een vorm van acute leukemie, vaak AML.
-
Atypische chronische myeloïde leukemie (aCML)
Dit is een erg zeldzaam ziektebeeld met bovendien een zeer verwarrende naam. Deze vorm van chronische leukemie heeft namelijk helemaal niets te maken met de bekende chronische myeloïde leukemie (CML). Het beeld lijkt er wel een beetje op, vandaar de naam atypisch, maar daar is ook alles mee gezegd. Deze vorm van leukemie is moeilijk te behandelen en lijkt nog het meest op de chronische myelomonocytenleukemie. De – voor CML – bekende middelen Glivec en alfa-interferon blijken ook niet te werken.
-
Juveniele myelomonocytenleukemie (JMML)
Van alle vormen van chronische leukemie is dit verreweg de zeldzaamste en deze wordt uitsluitend op de kinderleeftijd aangetroffen. Aangezien deze leukemie erg moeilijk te behandelen is, zullen patiëntjes bijna altijd een stamceltransplantatie ondergaan.
-
Behandeling van MDS
De behandeling van patiënten met MDS kan alle kanten op, variërend van helemaal geen therapie tot maximaal intensieve chemotherapie inclusief stamceltransplantatie. De keuze van de behandeling hangt af van veel verschillende factoren. Bij de behandeling is het daarom noodzakelijk precies te weten om welk type MDS het gaat.
MDS-type refractaire anemie of refractaire multilineage cytopenie
De beginvormen, zoals type refractaire anemie al dan niet met ringsideroblasten, behoeven vaak nauwelijks therapie omdat patiënten hier meestal weinig van merken. Mochten er wel klachten van de bloedarmoede zijn, dan is behandeling met af en toe een bloedtransfusie verreweg de simpelste manier. Behandeling met ijzer heeft geen zin, want er is geen tekort aan ijzer, maar er is eerder zelfs te veel ijzer in het lichaam. De anemie is veroorzaakt door een aanmaakstoornis die niet verholpen kan worden met ijzertabletten. De zeldzame RARS (refractaire anemie met ringsideroblasten) reageert een heel enkele keer goed op een hoge dosis vitamine B6 (pyridoxine).
Een enkele maal wordt gebruikgemaakt van groeifactoren zoals Epo (erytropoiëtine) en/of G-CSF. Zij kunnen worden gebruikt ter stimulering van de productie van gezonde rode en witte bloedcellen. De waarde van de behandeling met groeifactoren is omstreden. Een probleem is bovendien dat deze zeer dure medicamenten per injectie toegediend moeten worden en dat wanneer gestopt wordt met deze behandeling, het aantal rode en witte bloedcellen helaas weer snel terugloopt.
MDS-type refractaire anemie met excess aan blasten of varianten zoals CMML of atypische CML
Patiënten met deze drie groepen aandoeningen hebben over het algemeen een slechtere prognose. De behandeling hangt af van de leeftijd en conditie van de patiënt. MDS is met ‘gewone’ chemotherapie – zoals deze toegepast wordt bij ziekten als acute leukemie – moeilijk te behandelen. Bovendien komt de ziekte vaak voor bij ouderen, die zware chemotherapie slecht zullen verdragen. Recent is een nieuw middel – azacitidine (Vidaza) – beschikbaar gekomen dat erg goed werkzaam lijkt te zijn bij deze groepen patiënten. Vidaza wordt onder de huid gespoten (subcutaan toegediend) in een maandelijks schema van 7 dagen achtereen, gevolgd door 21 dagen rust. Omdat Vidaza beperkt houdbaar is, moet het – na aflevering door de apotheek – snel toegediend worden, wat vooralsnog thuisbehandeling bemoeilijkt. Vidaza is een medicijn dat in de cellen wordt ingebouwd en deze na een aantal celdelingen zal doden. Het effect laat daarom op zich wachten. Het duurt zeker 2 tot 3 maanden (2 à 3 kuren dus) voordat de werking zichtbaar wordt. Die werking zal zich uiten in een verbetering van het bloedbeeld en vaak ook in het verdwijnen van de blasten. Bij een klein deel van de patiënten lijkt het zelfs of de ziekte volledig verdwijnt.
Patiënten die niet in aanmerking komen voor Vidaza ondergaan een behandeling/begeleiding vooral gericht zijn op symptoombestrijding. Klachten die samenhangen met bloedarmoede worden zo nodig behandeld met bloedtransfusies. Voor infecties worden antibiotica gegeven, eventueel zelfs al als profylaxe (zie Selectieve darmdecontaminatie (SDD)).
Bij jongere patiënten (jonger dan 60 tot 65 jaar) wordt wél getracht met intensieve chemotherapie, eventueel gevolgd door een autologe (van de patiënt zelf) of allogene (van een donor) stamceltransplantatie alsnog genezing te bereiken. De intensieve chemotherapie is dezelfde als die bij acute myeloïde leukemie gegeven wordt. Bij jongere patiënten kan de toepassing van chemotherapie of stamceltransplantatie leiden tot een lange periode van vrij zijn van ziekteverschijnselen en soms ook tot volledig herstel.
MDS 5q- syndroom
Voor patiënten met het 5q- syndroom is binnenkort specifieke therapie beschikbaar. Het betreft het middel lenalidomide (Revlimid), dat in een lage dosis bij circa twee derde van de patiënten een goede verbetering geeft van het ziektebeeld. Patiënten ervaren een stijging van het bloedgehalte met het verdwijnen van de bloedarmoede; bloedtransfusies zijn dan niet meer nodig. Bovendien verdwijnt bij een deel van hen de specifieke chromosomenafwijking (verlies van een stukje van het vijfde chromosoom), wat suggereert dat lenalidomide echt iets doet aan de oorzaak van de ziekte.
Lenalidomide is al in Nederland beschikbaar als medicament voor de ziekte van Kahler (multipel myeloom). De verwachting is dat het ook snel voor de indicatie 5q- syndroom geregistreerd zal worden.

Dia 10
Conclusie
Het myelodysplastisch syndroom (MDS) is niet één ziektebeeld, maar een verzameling van beenmergziekten met als gemeenschappelijk kenmerk dat er fouten zijn in de aanmaak van bloedcellen. MDS kan betrekkelijk onschuldig zijn, maar kan zich ook ontwikkelen tot een ernstig ziektebeeld dat intensieve behandeling behoeft. De prognose hangt af van de leeftijd van de patiënt, het type MDS en de aan- of afwezigheid van specifieke chromosoomafwijkingen in de beenmergcellen.
Tot slot
Patiënten met MDS kunnen gebruikmaken van de activiteiten van de patiëntenvereniging voor patiënten met leukemie. Hier is voor gekozen, omdat sommige vormen van MDS beschouwd worden als een soort voorstadium van acute leukemie en patiënten wat therapie betreft dezelfde problemen kunnen ervaren als patiënten met acute of chronische leukemie. Bij deze patiëntenvereniging kunnen patiënten met alle verschillende soorten van MDS zich aanmelden. De vereniging is erg actief met vele voorlichtingsbijeenkomsten en een blad. De patiëntenvereniging bevindt zich binnen de overkoepelende patientenvereniging HEMATON voor patiënten met bloedkanker, lymfklierkanker, multipel myeloom en voor patiënten die een stamceltransplantatie hebben ondergaan.
Stichting Hematon
Postbus 8152
3503 RD Utrecht
Tel. 030-760 34 60
E-mail: secretariaat@hematon.nl
Website: www.hematon.nl