Inleiding
Primaire myelofibrose (PMF) behoort samen met essentiële trombocytemie (ET) en polycythemia vera (PV) tot de Philadelphiachromosoom-negatieve myeloproliferatieve aandoeningen (MPN). Het is een zeldzame aandoening met een geschatte incidentie van 0,5-1,5 per 100.000 inwoners per jaar. Bij 50-60% van de PMF-patiënten kan een JAK2 V617F-mutatie aangetoond worden, bij ongeveer 30% een CALR-mutatie (type I of type II) en bij 8% een MPL-mutatie. Ongeveer 12% van de patiënten heeft geen van deze 3 ‘driver’-mutaties (tripel negatief). Ook dient altijd chronische myeloïde leukemie (CML) te worden uitgesloten door het bepalen van een BCR/ABL-mutatie, ook in geval van de aanwezigheid van een ‘driver’-mutatie. De ziekte begint met een initiële prefibrotische/vroege fase (pre-PMF), waarbij er hypercellulair beenmerg is zonder of met minimale fibrose, en ontwikkelt zich in de tijd tot een fibrotisch stadium (overte PMF) met kenmerkend leuko-erytroblastair bloedbeeld met traandruppelcellen, splenomegalie of hepatosplenomegalie, LDH-verhoging en al of niet B-symptomen. Naast PMF wordt onderscheid gemaakt in secundaire of post-polycytemia vera-myelofibrose en post-essentiële-trombocytose-myelofibrose (post-PV MF, post-ET MF).
Diagnostiek
Welk onderzoek is nodig bij de diagnose PMF
- Anamnese met aandacht voor algemene klachten, constitutionele symptomen en klachten van splenomegalie (bij voorkeur ook objectiveren met Myeloproliferative Neoplasm-Symptom Assessment Form (MPN-SAF vragenlijst), bloedingsneiging, cardiovasculaire risicofactoren en jicht
- Lichamelijk onderzoek: bloeddruk, palpatie lever- en miltgrootte
- Bloedbeeld (inclusief leukocytendifferentiatie), reticulocyten, creatinine, leverenzymen, LDH, urinezuur, glucose en cholesterol/triglyceriden
- Mutatiebepaling: JAK2V617F-, CALR- en MPL-mutatieanalyse. Daarbij BCR-ABL1 genfusie (ter uitsluiting CML)
- Bloed- en beenmergmorfologie (bijvoorbeeld leuko-erytroblastair bloedbeeld, tear drop cells)
- Beenmergbiopt
- Cytogenetica (prognostische betekenis)
- Op indicatie:
- Aanvullende moleculaire diagnostiek middels NGS (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc.) bij patiënt met mogelijke indicatie allogene stamceltransplantatie (prognostische betekenis, MIPSS70/MIPSS70plus)
- In geval van verhoogde bloedingsneiging en/of indien trombocytose voorafgaand aan ingrepen met bloedingsrisico: sluit verworven Von Willebrand type II uit middels bepaling van Von Willebrand ristocetine-activiteit en antigeen (verstoorde ratio, met verminderde activiteit)
- Echo milt bij twijfel over splenomegalie (of bij adipositas)
Diagnostische ICC-criteria 2022
De 2022 ICC-classificatie maakt bij de diagnose myelofibrose onderscheid tussen pre-fibrotische myelofibrose (pre-PMF) en de ‘overte’ myelofibrose (overte PMF) (tabel 1).
| Pre-PMF# | Overte PMF# | |
|---|---|---|
| Beenmerg | Megakaryocytaire proliferatie en atypie, met toegenomen celrijkdom van het beenmerg (gecorrigeerd voor de leeftijd), granulocytaire proliferatie, en vaak verminderde erythropoëse | Megakaryocytaire proliferatie en atypie, granulocytaire hyper- of hypoproliferatie, verminderde erythropoëse |
| Reticulinefibrose | ≤ 1 | Reticuline- en/of collageenfibrose graad 2 of 3 |
| Exclusie | PV, ET, BCR-ABL1+ CML, MDS, of andere myeloïde neoplasie | PV, ET, BCR-ABL1+ CML, MDS, of andere myeloide neoplasie |
| Clonaliteit | Aantonen van een JAK2-, CALR-, or MPL-mutatie of indien deze mutaties niet aantoonbaar zijn een andere klonale marker* of afwezig zijn van reactieve oorzaken voor beenmergfibrose** | Aantonen van een JAK2-, CALR-, or MPL-mutatie of indien deze mutaties niet aantoonbaar zijn een andere klonale marker* of afwezig zijn van reactieve oorzaken voor beenmergfibrose** |
| Minor criteria |
|
|
| Voetnoten: # Diagnose: 3 major criteria in combinatie met minimaal 1 minor criterium. *: In de afwezigheid van de drie klonale markers, kan het bepalen van andere somatische mutaties behulpzaam zijn bij het aantonen van klonaliteit (bijvoorbeeld: ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc.). **: BM-fibrose secundair aan infectie, auto-immuunaandoening, chronisch inflammatoire aandoening, hairy-cell-leukemie of andere lymfatische neoplasie, gemetastaseerde ziekte of toxische beenmergafwijkingen. |
||
Klinische presentatie PMF
Meer dan de helft van de patiënten met PMF heeft constitutionele symptomen zoals vermoeidheid, gewichtsverlies, nachtzweten en/of koorts. Daarnaast kunnen er ook andere ziektegerelateerde klachten zijn zoals concentratiestoornissen, jeuk, botpijn en klachten gerelateerd aan de splenomegalie. Dit kan bij diagnose en follow-up vastgelegd worden met de MPN-SAF vragenlijst. Verder kan een verworven Von Willebrand-ziekte type II (VvWD) voorkomen bij extreme trombocytose maar ook bij lagere trombocytenaantallen. Een verminderde ratio van Von Willebrand-ristocetine-activiteit ten opzichte van Von Willebrand-antigeen is daarbij het meest voorspellend. Aangezien het risico op trombotische complicaties ook bij PMF afhankelijk is van cardiovasculaire risicofactoren dienen deze bij diagnose bepaald te worden.
Symptomen gerelateerd aan extramedullaire hematopoëse
- hepatosplenomegalie
- EMH buiten lever/milt: longen (kortademigheid; pulmonale hypertensie), pleura (pleuravocht), peritoneum (ascites), ruggenmerg (neurologische klachten).
Complicaties
- Portale hypertensie, met ascites, varicesbloedingen bij 7% van de patiënten
- Miltinfarct (hypodense laesie op CT)
- Extramedullaire hematopoëse: serosa-oppervlak (pleuravocht, ascites), longen, tractus urogenitalis (hematurie), paraspinale en epidurale ruimte (myelum of wortelcompressie). Diagnose met behulp van technetiumscan, MRI en/of biopt (cave bloeding)
- Ernstige bot-/spierpijn, gewrichtspijn
Prognose
Er zijn diverse scoringssystemen gepubliceerd om de prognose van PMF-patiënten te beoordelen. Deze scores zijn over het algemeen gebaseerd op retrospectieve data. Deze scoringssystemen zijn overigens niet gevalideerd voor de patiëntencategorie die ruxolitinib gebruiken of gebruikt hebben. Het advies is om de meest recente scoringssystemen te hanteren. De DIPSS-plus score is voor alle PMF-patiënten bruikbaar, de MIPSS70 en de MIPPS70plus score alleen voor patiënten van 70 jaar of jonger en kandidaat voor allo-SCT. Alhoewel de meeste scoringssystemen gevalideerd zijn voor PMF, worden deze in de praktijk ook gebruikt voor post-ET/PV MF. Echter, verschillende onderzoekers hebben aangetoond dat de voorspellende kracht van de DIPPS-plus score afneemt bij patiënten met post-ET/PV MF. Bij deze categorie van patiënten kan de MYelofibrosis SECondary to PV and ET Myelofibrosis (MYSEC-PM) score gebruikt worden. De effectiviteit van dit prognostisch model is bevestigd door verschillende onderzoeksgroepen. Voor patiënten met PMF of post-ET/PV MF die kandidaat zijn voor stamceltransplantatie kan de MTSS score aanvullend gebruikt worden om de overlevingskans na transplantatie in te schatten.
Primaire myelofibrose
- Inschatten van “overall survival” op basis van: Dynamic International Prognostic Scoring System Plus (DIPSS Plus) (tabel 2)
- Inschatten van “overall survival” ter identificatie van kandidaten voor allogene SCT: MIPSS70 (tabel 3) or MIPSS70plus; versie 2 (tabel 4)
Post-ET/PV Myelofibrose
- Inschatten van “overall survival” op basis van: MYelofibrosis SECondary to PV and ET Myelofibrosis Prognostic model (MYSEC-PM; tabel 5).
Stamceltransplantatierisico voor PMF of Post-ET/PV Myelofibrose
- Inschatten “overall survival” na stamceltransplantatie op basis van MTSS (tabel 6).
| DIPSS+ parameter | > Score |
|---|---|
| Leeftijd > 65 jaar | 1 |
| Constitutionele symptomen | 1 |
| Hb < 6,2 mmol/l | 1 |
| Leukocytose > 25 × 109/l | 1 |
| Blasten perifeer bloed > 1% | 1 |
| Trombocytopenie < 100 × 109/l | 1 |
| Cytogenetica: complex, +8,-7/7q-, i(17q), 5/5q-, 12p-, inv(3) of 11q23 rearrangement | 1 |
| Erytrocytentransfusie afhankelijkheid | 1 |
| Risico-indeling DIPSS Plus | Score | Mediane Overall survival (maanden) |
| Laag | 0 | 185 |
| Intermediair-1 | 1 | 78 |
| Intermediair-2 | 2-3 | 35 |
| Hoog | ≥4 | 16 |
Tabel 2. DIPSS Plus
| MIPSS70 parameter | Score |
| Beenmergfibrose graad 2 of 3 | 1 |
| Constitutionele symptomen | 1 |
| Hb < 6,2 mmol/l | 1 |
| Leukocytose > 25 × 109/l | 2 |
| Blasten perifeer bloed > 2% | 1 |
| Trombocytopenie < 100 × 109/l | 2 |
| Hoog moleculair risico: 1HMR afwijking* | 1 |
| 2 of meer HMR-mutaties* | 2 |
| Afwezigheid CALR-type 1 mutatie | 1 |
| *ASXL1, SRSF2, IDH1/2, EZH2 (U2AF1) | |
| Risico indeling MIPSS70 | Score | Mediane Overall survival (jaren) |
| Laag | 0 | 22,7 |
| Intermediair | 2-4 | 7,1 |
| Hoog | ≥ 5 | 2,3 |
Tabel 3. MIPSS70
| MIPPS70+ parameter | Score |
| Constitutionele symptomen | 2 |
| Hb < 5,6 mmol/l (mannen); Hb < 5 mmol/l (vrouwen) | 2 |
| 5,6 > Hb < 6,8 mmol/l (mannen); 5 > Hb < 6,8 mmol/l (vrouwen) | 1 |
| Blasten perifeer bloed ≥ 2% | 1 |
| Hoog moleculair risico: 1 HMR afwijking* | 2 |
| 2 of meer HMR-mutaties* | 3 |
| Afwezigheid CALR-type-1 mutatie | 2 |
| Ongunstige karyotype ** | 3 |
| Zeer hoog risico karyotype *** | 4 |
| *ASXL1, SRSF2, IDH1/2, EZH2, U2AF1 **Ieder afwijkend karyotype, behalve: 20q-, 13q-, +9, chromosoom 1 translocatie/duplicatie, -y of geslacht chromosoomafwijking anders dan –y *** single/multiple afwijkingen van -7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, of andere autosomale trisomie behalve +8/+9 (e.g. +21, +19) |
|
| Risicoindeling MIPSS70+ (versie 2) | Score | Mediane 10-jaarsoverleving |
| Zeer laag | 0 | Niet bereikt |
| Laag | 1-2 | 16,4 |
| Intermediair | 3-4 | 7,7 |
| Hoog | 5-8 | 4,1 |
| Zeer hoog | ≥ 9 | 1,8 |
Tabel 4. MIPSS70plus; versie 2
| MYSEC-MF parameter | Score |
| Leeftijd | 0,15 per jaar |
| Trombocytopenie < 150 × 109/l | 1 |
| Hb < 6,8 | 2 |
| Blasten perifeer bloed > 2% | 2 |
| Constitutionele symptomen | 1 |
| Afwezigheid CALR-mutatie | 2 |
| Risico-indeling MYSEC-MF | Mediane overall survival (jaar) |
| Laag | Niet bereikt |
| Intermediair-1 | 9,3 |
| Intermediair-2 | 4,5 |
| Hoog | 2 |
| Voor risicocalculatie gebruik de calculator: http://www.mysec-pm.eu/ | |
Tabel 5. MYSEC-MF
| MTSS parameter | Score |
| Leukocytose > 25 × 109/l | 1 |
| Trombocytopenie < 150 × 109/l | 1 |
| Karnofsky performancestatus | 1 |
| Afwezigheid CALR- of MPL-mutatie | 2 |
| Leeftijd > 57 jaar | 1 |
| HLA-mismatched unrelated | 2 |
| ASXL1-mutatie aanwezig | 1 |
| *ASXL1, SRSF2, IDH1/2, EZH2 | |
| Risico indeling MTSS | Score | 5 jaar overleving (%) |
| Laag | 0-2 | 83 |
| Intermediair | 3-4 | 64 |
| Hoog | 5 | 37 |
| Zeer hoog | 6-9 | 22 |
Tabel 6. MTSS
Behandeling
Filter trials en protocollen voor dit ziektebeeldBij PMF en secundaire MF is allogene stamceltransplantatie (allo-SCT) de enige curatieve behandelingsmodaliteit. Bij allo-SCT in MF moet de post-transplantatie mortaliteit en morbiditeit (GVHD) afgewogen worden tegen de bekende mediane OS met natuurlijk het risico op ontwikkeling van sAML. Figuur 1 schetst een operationeel behandelingsalgoritme dat is gebaseerd op risicostratificatie volgens MIPSS70+; versie 2.0. Bij patiënten waarbij geen cytogenetische analyse mogelijk is kan het risico ingeschat worden door middel van de MIPP70 score. Bij patiënten waarbij geen NGS gedaan is, is de DIPSSplus score het meest geschikt om de overleving en indicatie voor allo-SCT in te kunnen schatten.
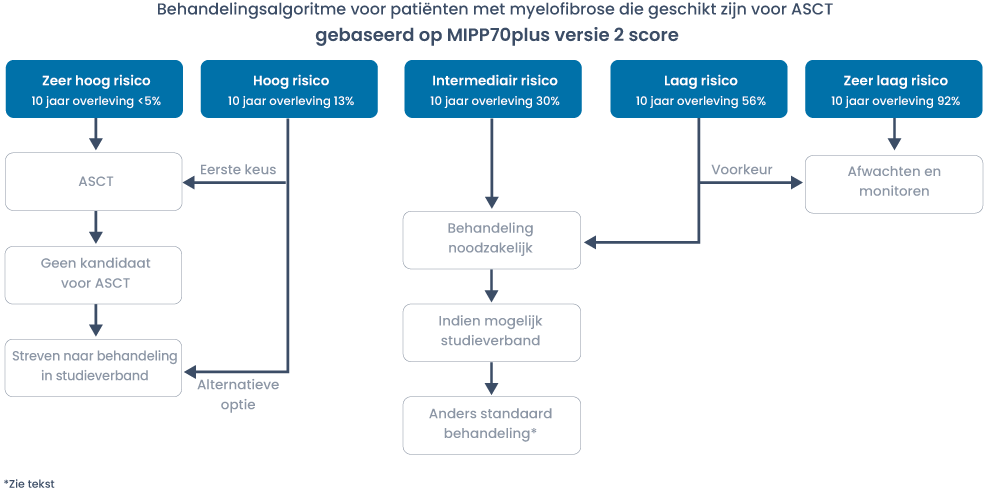
Voor verdere uitwerking van de behandelmogelijkheden zie de daarop volgende tekst.
Indicatie starten behandeling
- Leeftijd > 60 jaar, streef trombocytenwaarde < 400 × 109/l. (Doorgemaakte) trombo-embolische complicatie, streef trombocytenwaarde < 400 × 109/l
- Verworven Von Willebrand ziekte (VvWD), streef verlagen trombocyten en/of remissie VWD
- Symptomatische splenomegalie
- PMF-gerelateerde symptomen ter verbetering QoL (MPN-SAF score vervolg)
- Progressieve myeloproliferatie, streef naar leukocyten < 10-15 × 109/l
1e lijn
- Indien symptomatische splenomegalie en/of trombocytose/leukocytose: hydroxycarbamide, start dosering 1dd 500-1000 mg oraal (aandachtspunten bij start Hydrea: zie tabel 7) of gePegyleerd interferon α-2a (Pegasys®), start dosering 30-45-90 microgram/week s.c. (aandachtspunten bij start Pegasys: zie tabel 7)
- Indien ernstige PMF-gerelateerde symptomen (maak hiervoor gebruik van MPN-SAF vragenlijst): ruxolitinib. Start dosering 2dd 20 mg, afhankelijk van trombocytenwaarde startdosering aanpassen (aandachtspunten bij start ruxolitinib in tabel 7)
2e lijn
- Indien symptomatische splenomegalie en/of PMF-gerelateerde symptomen: Ruxolitinib. Start dosering 2dd 20 mg, afhankelijk van trombocytenwaarde startdosering aanpassen. Aandachtspunten bij start ruxolitinib zijn weergegeven in tabel 7
- Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Gepegyleerd interferon α-2a (Pegasys®), start dosering 30-45-90 microgram/week s.c.
- Combinatie van behandelingen
- Indien alleen trombocytose: anagrelide, start dosering 2dd 0,5 mg, iedere week te verhogen met 0,5 mg/dag extra op geleide van trombocytenaantal. Maximale dosis: 10 mg/dag en 2,5 mg/gift.
Aanvullende maatregelen
- Trombocytenaggregatieremming (tenzij forse trombopenie of bloedingsneiging): Acetylsalicylzuur 80 mg/carbasalaatcalcium 100 mg per dag, liefst ’s avonds inname. Indien VG met DVT dan over op OAC i.p.v. trombocytenaggregatieremming
- Uraatsteen en jicht profylaxe indien verhoogd urinezuur, of jicht in voorgeschiedenis; allopurinol 1 dd 300 mg
- Goede behandeling van aanvullende cardiovasculaire risicofactoren
Anemie
- Androgenen: danazol 2 dd 1-2 caps. van 100-200 mg
- Darbopoietine 150-500 mcg/1-3 wk afh. van epospiegel
Niet-responsief extramedullaire hematopoëse (EMH; excessieve myeloproliferatie (MP))
- Melfalan (2 mg/tab), 2-3 ×/week 2 mg; respons na ca. 6-7 mnd bij 99 patiënten: met name leukocytose, trombocytose (86 en 93%), splenomegalie: 23% CR, 32% > 50% afname in grootte; anemie: 60% verbetering, 38% werd transfusie onafhankelijk; 3/8 afname fibrose; responsduur: 2-2,5 jaar.
- Radiotherapie:
- Milt: alleen indien operatie is gecontra-indiceerd. Respons op bestraling is goed maar aanzienlijke myelosuppressie. Bestraling (2,5-5,0 Gy/dag, ged. 4-5 dagen) leidt in 95% tot verbetering van klachten en verkleining van de milt (gemiddeld 5 cm, cranio-caudaal) voor de duur van ca. 6 mnd. Bij 44% duidelijke cytopenieën, bij 26% langdurige ernstige cytopenie; bij de helft hiervan fatale sepsis of bloeding; de overall mortaliteit is 13% (Mesa et al., 2006).
- Lever/buik: in verband met hepatomegalie, ascites.
- Longen: in verband met EMH, pulmonale hypertensie.
- Spinaal: in verband met EMH en compressie ruggenmerg.
- Botpijn: veelal bij leukemische transformatie.
| Aandachtspunten bij Ruxolitinib, Peg-INFa en Hydrea |
| Ruxolitinib |
| Medicatie niet abrupt staken maar de dosis in 2 weken verminderen ter preventie van “withdrawal syndroom”. |
| Voorafgaand aan het starten van ruxolitinib overwegen of screening op tuberculose zinvol is. |
| Standaard antimicrobiële of virale profylaxe niet geïndiceerd. Patiënten voorlichten ten aanzien van verhoogd risico op herpes zoster-reactivatie. |
| Geneesmiddeleninteractie met sterke/matige CYP3A4-remmers of tweevoudige remmers van CYP2C9 en CYP3A4. Voorbeelden: claritromycine, ciprofloxacin, azolen, verapamil. |
| Hydrea |
| De voornaamste kortetermijnbijwerkingen zijn megaloblastaire anemie, leukopenie, stomatitis, been ulcera, acne, maagpijn en diarree, wat bij 10-15% van de patiënten leidt tot het staken van de medicatie. Cave zonlicht met meer kans op huidmaligniteiten, m.n. op de langere termijn. Ook meer kans op secundaire maligniteiten op de langere termijn. |
| Bij starten Hydrea frequentere controle noodzakelijk i.v.m. ernstige cytopenieën, ook bij nierinsufficiëntie is deze kans groter. |
| Gepegyleerd interferon α-2a |
| Geriatrische patiënten: Neuropsychiatrische (wo. depressie en epilepsie), cardiale (ritme st.) en systemische (griepachtige) bijwerkingen kunnen ernstiger zijn. Start low en go slow geeft minder bijwerkingen en verhoogt de kans op acceptatie medicijn voor een langetermijnbehandeling. |
| Patiënten met nierinsufficiëntie: de dosis PEGASYS moet worden verlaagd bij patiënten met een creatinineklaring van minder dan 30 ml/min. |
| Gecontra-indiceerd bij: 1) auto-immuunziekten; 2) ernstige reeds bestaande hartaandoeningen (zoals een instabiele hartaandoening of een hartaandoening die niet onder controle is in de voorgaande 6 maanden); 3) Aanwezigheid of voorgeschiedenis van een ernstige psychiatrische aandoening (ernstige depressie, suïcidaal gedrag), epilepsie is een relatieve ci. |
Tabel 7. Aandachtspunten bij Ruxolitinib, Peg-INFa en Hydrea
Literatuurlijst
- Arber DA, et al. International Consensus Classification [ICC] of Myeloid Neoplasms and Acute Leukemias. Blood 2022;140:1200.
- Cervantes et al. Blood. 2009;113:2895.
- Passamonti et al. Blood. 2010;115:1703.
- Gangat et al. JCO. 2011;29:392.
- Guglielmelli et al. J Clin Oncol. 2018;36:310.
- Tefferi et al. JCO. 2018;17:1769.
- Gagelmann et al. Blood. 2019;133:2233.